Something Old... Something Blue
- Stanley Scheindlin, DSc
In 1906, Alois Alzheimer, together with Emil Kraepelin and two Italian colleagues, studied the brain of a deceased patient suffering from profound short-term memory loss and dementia. They observed the cerebral cortex was smaller than normal and identified, in stained sections, fibrous aggregates of protein, more commonly called amyloid plaques. Later that year, Alzheimer presented the first evidence of observable brain pathology associated with malfunctioning cognition and dementia, yet it was Kraepelin who first referred to the pathophysiology as Alzheimer’s disease (AD) (1).

Alois Alzheimer
An estimated 26.6 million people worldwide have AD; this number may quadruple by 2050 (1); in the US alone, there are thought to be 4.5 million afflicted patients (2). Establishing a therapy that would delay the onset of AD by only one year would save $9 billion in the US alone (3).
Recently, results from a clinical trial, presented at the 2008 International Conference on Alzheimer’s Disease (ICAD), showed that an investigational drug was capable of arresting progression in mild and moderate AD (4, 5). Enthusiasm must be tempered, as the study involved relatively few patients and the results must be confirmed by more extensive trials. Nevertheless, this was the first intervention ever to arrest the inexorable progression of AD.
On reading this news, the first question to come to mind was: what is the identity of the investigational drug? The answer came as a stunning surprise. Not a bioengineered protein, not a monoclonal antibody, not a new synthetic compound specially designed to fit snugly into an enzyme’s pocket, the drug candidate is a dye known since 1876: methyl-thioninium chloride, or methylene blue (MB).
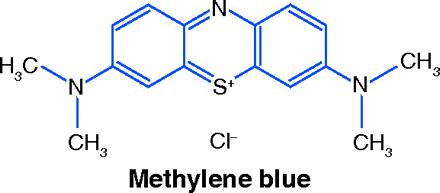
Despite a long and varied history, MB has fallen into obscurity. Its rebirth might come soon: if further clinical research yields favorable results, MB may become a compound of surpassing interest to scientists and lay persons alike. It therefore seems appropriate to review its 130-year medical history and explore the rationale for its possible effectiveness against AD.
Early History of Methylene Blue
Methylene blue is made by reacting N,N-Dimethyl-p-phenylenediamine in acidic medium with hydrogen sulfide and ferric chloride. The sulfur atom bridges two molecules of the phenylenediamine, forming a phenothiazine, and is oxidized by the ferric ion to a positively charged thionium ion. For industrial use as a dye, MB is supplied as a double chloride with zinc; for bacteriologic and medicinal uses the zinc-free form is employed.
First synthesized in 1876, concomitant with the beginnings of bacteriology, MB found immediate use in the staining of microorganisms, and its effect on bacterial metabolic behaviors was also studied. MB became an early research interest of Paul Ehrlich, who would later participate in developing diphtheria antitoxin, concocting the first chemotherapy for syphilis (salvarsan), and enunciating the receptor theory of antibody action. Ehrlich made the important observation that in some tissues, MB was reversibly reduced to a colorless (leuco-) form He also found that it possessed a selective affinity for nerve cells and nerve fiber endings, a finding which may have significance in today’s context. Reasoning that it might therefore interfere with conduction of pain impulses by the nerves, Ehrlich obtained a supply of purified MB and, together with a physician named Lippmann, conducted a clinical trial in patients with neuralgia and arthritis (6). MB produced an analgesic effect but it tended to damage the kidneys if used continuously (7).

Paul Ehrlich
MB as an Antimalarial
Noting that malaria-causing Plasmodia were stained by MB and that the dye seemed safe for short-term administration, Ehrlich and Guttmann, in 1891, administered MB as a treatment for malaria, on two patients in a Berlin hospital. The results were positive, though not equal to those with quinine (6). Still, the desirability of another antimalarial, lacking quinine’s side effects and usable by patients unable to tolerate quinine, led to further trials by other physicians. In 1904, Horatio C. Wood, Jr. performed what today would be called a meta-analysis of the clinical results published as of that date. Reviewing 425 cases (including Ehrlich’s two) gleaned from eleven published reports, he concluded that 362 (85%) were final cures, whereas thirty-one suffered relapses (8).
Anopheles gambiae, bearer of malaria
Although the search for antimalarials focused on synthetic substitutes for quinine, physicians still turned to MB under certain conditions. Thus, a report from World War II era Soviet Union described the treatment of forty patients with a type of malaria that had proved refractory to repeated courses of quinine. Treatment consisted of five or six intravenous (iv) infusions of 15–20 mL of 1% MB in 25% glucose. In most of these patients, fever was terminated after the first or second infusion and attacks did not recur. In twenty of the thirty-five patients in whom plasmodia had been seen, the organisms disappeared from the peripheral blood. The author concluded that MB has a definite role when the patient is quinine-intolerant or does not respond to quinacrine and plasmoquine (9).
More recently, the antiplasmodial activity of MB was confirmed in vitro by Vennerstrom et al. whereby several dyes were tested against three different strains of P. falciparum, each having a different drug susceptibility/resistance profile. Dyes of the xanthine, azine, oxazine, and thiazine categories were tested. The thiazine (phenothiazine) dyes were found to be the most active antimalarials while showing low cytotoxicity MB was among the most potent of this group (10).

Plasmodium falciparum
MB and Antimalarial Synthesis
MB’s main contribution to the cure of malaria lies not in its direct use in therapy, but in serving as a template for the synthesis of substitutes for quinine. Sneader (11) relates that Ehrlich had suggested making derivatives of MB as possibly improved antimalarials. He was prevented from pursuing his idea, first, because no animal model for malaria existed at the time on which compounds could be tested, and second, because he became involved in research on diphtheria antitoxin [see (12) for historical details of mushing the antitoxin to Nome].
Only in 1924 did a screening method, using canaries, become available. Werner Schuleman and colleagues at I. G. Farben then took up Ehrlich’s suggestion. By substituting a diethylaminoethyl (DEAE) side chain for one of MB’s methyl groups, they obtained a promising compound, but as it was deeply colored, they felt it might be unacceptable to patients. Schuleman’s group substituted quinoline for the phenothiazine nucleus, retained the DEAE side chain, and added some features of the quinine structure. Of the hundreds of compounds prepared, the first clinically tested and marketed compound was Plasmoquine (pamaquine). Thus, the first synthetic drug designed as an antimalarial derives partly from MB (11).
A Urinary Antiseptic Prescription
Because it possesses antibacterial activity and is secreted into the urine, MB is a key ingredient in a combination product for treating infections and painful disorders of the urinary tract. This product is remarkable for its longevity: it predates the New Drug Application requirement of the 1938 Food, Drug, and Cosmetic Act, and is still on the market today.
Originating in an era when physicians were accustomed to writing multi-ingredient prescriptions (termed polypharmacy), the product resembles such a prescription. In addition to 5.4 mg of MB, each tablet contains atropine sulfate 0.03 mg, hyoscyamine 0.03 mg, methenamine 40.8 mg, benzoic acid 5.4 mg, and phenyl salicylate 18.1 mg (13). The atropine and hyoscyamine, both parasympatholytic alkaloids, were intended to relieve the pain of smooth muscle spasms in the urinary tract. Methenamine, a complex compound made by condensing formaldehyde with ammonia, was at one time highly regarded. In acid medium methenamine breaks down into its components; thus, if the urine is acidified (e.g., by benzoic acid) the antibacterial action of free formaldehyde would be released. In the 1930s, methenamine was recommended for many conditions, including pyelitis, cystitis, prostatitis, urolithiasis, gonorrhea, meningitis, and polio (14) truly a wonder drug, had it worked! The rationale for adding phenyl salicylate (salol) resided in its antiseptic action, as it contains insufficient salicylate for any analgesic effect. This formula is on the market as Urised and Uriseptic, and in generic form as Urinary Antiseptic #2 (13).
Treatment of Methemoglobinemia
MB is used therapeutically for a relatively uncommon condition wherein high concentrations of methemoglobin are present in the blood. Methemoglobin, an oxidation product of hemoglobin in which the iron atom is in the ferric (trivalent) state, gives the blood a brown color but does not combine with oxygen, thus reducing the oxygen-carrying capacity of the blood. Methemoglobinemia may be congenital, or may be acquired, for example, from well-water containing nitrites or from drugs such as benzocaine and sulfonamides. MB acts through the MB leuco-MB redox system. Upon iv injection MB reacts with reductases in erythrocytes, forming its dihydro derivative, colorless leuco-MB, which can reduce methemoglobin back to normal hemoglobin (15).
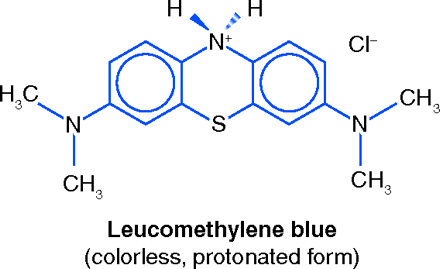
This use of MB was discovered long ago, and was well established by the 1940s. In 1947, Bodansky and Gutmann induced varying degrees of methemoglobinemia severity in dogs. By correlating their animal data with results published in the clinical literature they confirmed that an iv dose of 1–2 mg/kg of MB is rapidly effective against the symptoms of severe methemoglobinemia (16). In 1993, Eldadah and Fitzgerald recommended that methemoglobinemia be treated by iv administration of 1–2 mg/kg of MB as a 1% solution, infused over fifteen minutes. A second dose may be given, up to a maximum of 7 mg/kg/24 hrs (17). Methylene Blue Injection, a 1% sterile solution of MB, is officially recognized in the United States Pharmacopeia (USP).
Drug Treatment of AD: Present and Future
Currently Approved Drugs
The drugs currently used to treat AD patients act on brain neurotransmitters, although it is recognized that they do not address the cause. Some of the signs and symptoms of AD can be attributed to a deficiency of cholinergic neurotransmission part of the cholinergic system theory of AD. Thus, enhancing cholinergic function by inhibition of cholinesterase could be helpful. Four AD drugs based on this mode of action have been developed. Cholinesterase inhibitors do not alter the course of the dementia but act to slow cognitive demise. As AD progresses and fewer cholinergic neurons remain functional, the effect of these drugs diminishes (18).
The first centrally acting cholinesterase inhibitor to become available was tacrine (CognexR). Its clinical effect was small, and was further limited by its large intra-individual variation in oral bioavailability and its potential liver toxicity, as evidenced by an increase in blood liver enzymes in about 50% of patients (19).
Three additional cholinesterase inhibitors are approved in the United States. The oldest, donezepil (AriceptR), has been in use since 1997. Rivastigmine (ExelonR) also acts by the same mechanism. It is structurally related to physostigmine, an alkaloid first isolated in 1864 from Calabar beans. Physostigmine is a potent cholinesterase inhibitor, administered by injection or by mouth, but it is too short-acting to be very useful (20). ExelonR, in capsules and oral solution, must be taken twice daily. Recently, an ExelonR transdermal patch has become available, requiring application only once per day. The final approved cholinesterase inhibitor of plant origin is galantamine (RazadyneR). Isolated in the 1950s from Caucasian snowdrops and from species of Narcissus, its structure and methods of synthesis were quickly worked out. In immediate release form it must be taken twice a day. A once-daily capsule (RazadyneR ER) is also available (21, 22).

Calabar Beans

Caucasian Snowdrops
Memantine (NamendaR), a non-cholinesterase inhibitor, is approved for AD and antagonizes N-Methyl-d-aspartate receptors (NMDARs). Persistent activation of NMDARs in the central nervous system (CNS) by glutamate contributes, according to one hypothesis, to symptoms of AD. Memantine acts by binding to NMDAR-operated cation channels (23).
Methylene Blue to the Rescue?
As noted, none of the above drugs attacks the cause of AD, which is believed to involve brain proteins and peptides rather than small-molecule neurotransmitters. Autopsy studies have revealed that the brains of patients sufering from the dementia of AD exhibit an accumulation of plaques, which are deposits of the peptide beta-amyloid, and neurofibrillary tangles (NFTs), composed of abnormal microtubules (24, 25). The number of tangles correlates more closely with neuron loss and dementia than do the numbers of plaques (26). It is not known whether the plaques and NFTs cause AD, are inactive by-products of the disease process, or are actually protective. There has been considerable research effort aimed at dissolving beta-amyloid plaques or preventing their accumulation, but thus far, no potential therapeutic agent has emerged.
Another school of thought posits that the NFTs are a more appropriate target. The microtubules which constitute the tangles are composed of a polypeptide, tubulin, and a heat-stable “tubule assembly protein,” named tau, which catalyzes the polymerization of tubulin into microtubules (27). Tau protein has been suggested as a therapeutic target for drugs intended to combat AD. A significant study was recently carried out in a mouse model of AD. Mice genetically engineered to develop beta-amyloid plaques and age-related AD-like symptoms were crossed with mice in which the tau gene had been inactivated, resulting in offspring with lower levels of tau. In these mice a reduction of endogenous tau by as little as 50% prevented memory loss and other AD symptoms (28). An earlier in vitro study showed MB blocks the tau-tau interaction that would lead to the formation of NFTs, whereas other phenothiazines (i.e., neuroleptic drugs such as chlorpromazine or fluphenazine) were inactive in this respect (29). The principal investigator of this study is also a principal in the start-up firm that conducted the clinical trial on MB cited at the beginning of this article (1).
Will MB prove to be the first medicine to make a real difference in AD? There are formidable obstacles. The failure rate is high in clinical Phase III among compounds which pass Phase II successfully. The extent to which MB crosses the blood-brain barrier is unknown. Further, its rapid excretion via the urine and the absorption problem hinted at in the ICAD report point to additional potential difficulties. Thus, there are good reasons for expectations to be muted. Nevertheless, if its early promise in AD should be fulfilled, MB would not be the first medicinal to come back from obscurity. Arsenic trioxide, in use for centuries but deemed worthless in the second half of the twentieth century, is now approved for a rare form of leukemia. Thalidomide, the poster-child of disastrously dangerous drugs, has been rehabilitated as an orphan drug for multiple myeloma and erythema nodosum leprosum, a painful inflammatory complication of leprosy. A more benign example is provided by the vitamin folic acid, which can mask pernicious anemia and hamper its timely diagnosis. This finding, in 1947, prompted folic acid’s exclusion from multivitamin products. For about thirty years, the vitamin was relegated to insignificance, until research in the 1980s and 1990s established that it prevented birth defects such as spina bifida. As a result, fortification of all enriched grain products with folate has been a government-mandated requirement since 1998.
MB has been in use as a medicinal, to a greater or lesser extent, continuously throughout the past century. It may now return to play an important therapeutic role, either per se or as a structural model for the synthesis of more effective molecules to combat AD.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Stanley Scheindlin, DSc, holds a BS in pharmacy from Temple University and graduate degrees in pharmaceutical chemistry from Philadelphia College of Pharmacy and Science (now University of Sciences in Philadelphia). His academic research dealt with plant constituents and chemical interactions of vitamins. In his pharmaceutical industry career, he handled new drug formulation developments, and later regulatory affairs, presiding over the filing of about 100 generic new drug applications and two innovative drug applications. Now retired, his activities include volunteer work, consulting, and writing Reflections pieces for this journal. E-mail stansch{at}verizon.net

