Let’s Go Rafting: Ligand Functional Selectivity May Depend on Membrane Structure
One of the most exciting developments in the area of G protein–coupled receptor pharmacology in the twenty-first century has been the maturation and wide acceptance of the concept of ligand functional selectivity, variously known as biased agonism, conformational selection, agonist-directed signaling, relative activity, differential engagement, or stimulus trafficking (1, 2). In essence, functional selectivity occurs when agonists display different potency, efficacy, or both for different responses mediated through a single type of receptor.
The activation of a receptor can lead to stimulation of different intracellular signaling events, such that:

where, according to traditional receptor theory, the relative degree of activation of E1 and E2 is the same for every agonist. Under the concept of functional selectivity, some ligands, acting at a single receptor, may preferentially activate E1 whereas other ligands, acting at the same receptor in the same system, may preferentially activate E2, as follows:

where affinity and efficacy for different agonists can be dissociated to produce functionally selective responses. Apparent functional selectivity can be observed in different systems or cell types where alterations in the strength of signal can be accounted for by differences in receptor levels or agonist partial efficacy. True functional selectivity cannot be accounted for by a single receptor active-state but involves multiple agonist-dependent receptor active states (3).
Functional selectivity can be observed with respect to the activation of different signal transduction responses. It can also be observed with respect to the ability of different ligands to produce receptor regulation events such as desensitization and internalization (4). Developments in this area have been particularly interesting and somewhat controversial, with respect to the opiate receptor system (5).
A recent study (6) provides evidence that one of the factors that determines differential coupling of the μ-opiate receptor (MOR) is its location within different domains of the cell membrane. The MOR is the main high-affinity binding site for morphine and similar opioid compounds. Agents that activate the MOR produce analgesia, euphoria, and constipation by actions in the peripheral, central, and enteric nervous systems. MOR activation leads to several changes in cells, including closure of K+ channels, inhibition of adenylyl cyclase (AC), and activation of extracellular signal–regulated kinase (ERK) (7).
Functional selectivity has been demonstrated for several aspects of MOR signaling. For example, in HEK293 cells, agonist ligands (d-Ala2, N-Me-Phe4, Gly5-ol)- enkephalin (DAMGO) and etorphine lead to receptor endocytosis, while morphine does not (8). Also, some opiate agonists inhibit AC more effectively than they increase ERK phosphorylation (9). This latter difference may result from differences in coupling to G proteins or to β-arrestin (10). Zheng et al. had previously shown that morphine activates ERKs via the G protein–dependent pathway but not through the β-arrestin–dependent pathway. In contrast, etorphine activated ERKs in a β-arrestin–dependent manner (9).
Recent data suggest that this is partly controlled by the membrane environment, in particular membrane rafts. Membrane rafts are defined as “small (10–200 nm), heterogeneous, highly dynamic, sterol- and sphingolipid-enriched domains that compartmentalize cellular processes” (11). Membrane rafts participate in GPCR localization within the membrane (12) and compartmentation of cell signaling via colocalization of signaling components (13).
In HEK293 cells stably expressing a hemagglutinin-tagged MOR and in endogenous MOR-expressing hippocampi dissected from mouse brains, membrane raft and non-raft domains were separated by sucrose gradient centrifugation. In the absence of agonist, MOR were located within the membrane raft. Etorphine, but not morphine, led to translocation of MOR to non-raft domains. Under control conditions, 75% of MOR was co-localized with a marker of membrane rafts, as shown by confocal microscopy. When morphine was applied, this percentage remained the same. When etorphine was applied, only 30% of the receptor fluorescence co-localized with the raft marker, suggesting that morphine and etorphine lead to distinct receptor states and that the etorphine-induced conformation preferentially localizes to the nonraft domains.
Treatment of MOR-expressing HEK293 cells with methyl-β-cyclodextrin (to disrupt the integrity of the rafts) led to translocation of MOR and Gαi2 to nonraft domains. This disruption also inhibited the morphine-induced (G protein–dependent) but not etorphine-induced (β-arrestin dependent) ERK phosphorylation. The ability of both morphine and etorphine to inhibit forskolin-stimulated AC activity was decreased, owing in part to decreases in both the apparent affinities and efficacies of the agonists. These data suggest that location of the receptors and G proteins in membrane raft domains is required for G protein–dependent signaling. But what determines whether MOR is localized in raft or nonraft regions?
Coupling proteins appear to control the raft or nonraft location of MOR. Increasing cellular levels of Gαi2 attenuated MOR translocation to the nonraft domain in response to etorphine and attenuated ERK phosphorylation in response to etorphine, but increased AC inhibition by etorphine. Conversely, decreasing Gαi2 led to a greater fraction of MOR being located in nonraft domains in the absence of agonist, increased translocation of MOR into nonraft domains in response to both morphine and etorphine, increased ERK phosphorylation in response to these agonists, and decreased their ability to inhibit AC.
Furthermore, a direct interaction between the MOR and Gαi2 appears to exist, even in the absence of agonist, keeping MOR in the membrane raft domain. If the MOR-Gαi2 interaction was disrupted by deletion of the G protein–interaction motif of the receptor, the receptor was found in non-raft domains, but Gαi2 remained in raft domains. In this case, G protein–dependent inhibition of AC and activation of ERK by morphine were lost, but ERK activation by etorphine remained.
At the same time, it appears that MOR interaction with β-arrestin leads to translocation to nonraft domains. In cells taken from β-arrestin knockout mice, etorphine did not induce translocation of MOR, etorphine-induced ERK phosphorylation was decreased, and etorphine-induced AC inhibition was increased. This translocation is independent of β-arrestin induced internalization, but it does require Gαi2 dissociation (Figure 1⇓). Taken together, these results suggest that Gαi2, which is localized to the membrane rafts, controls the raft vs nonraft location of the MOR and are consistent with a preferential signaling of the MOR to Gαi2 in the raft domain and to ERK in the nonraft domains. The binding of morphine leads to a receptor conformation that has low affinity for β-arrestin and, upon hydrolysis of GTP by Gαi2, will rebind morphine and stay in the raft domain. The binding of etorphine leads to MOR-Gαi2 dissociation, allowing the MOR to diffuse to nonraft domains, but in this case the receptor conformation has a higher affinity for β-arrestin, will stay in the nonraft domains, and will activate ERK. These data also suggest that the cellular levels of Gαi2 will control signaling to AC or ERK. In cells with low amounts of Gαi2, the ERK signaling will predominate because the MOR will not be located in membrane rafts.
From a therapeutic perspective, one of the most intriguing implications of functional selectivity is that it may be possible to develop ligands to preferentially produce one or the other effects. If one effect is associated with a therapeutic effect (relief of pain in the case of opiates) and the other with side effects (tolerance and dependence), ligands could be developed to have more therapeutic value with fewer side effects. Also, changes in the distribution of signaling molecules in different membrane domains with age, disease, or drug history, may contribute to variations in signaling and drug effects. Other mechanisms that promote heterodimerization of opioid receptors and enable trafficking of such multimers to the cell surface may prove useful as therapeutic targets, as discussed by van Rijn and Whistler elsewhere in this issue of Molecular Interventions (14).
How the above results relate to the therapeutic actions of opiates are not clear, but a recent study of the β-adrenergic receptor (β-AR) suggests that there may be therapeutic advantages to developing ligands with functional selectivity. Carvedilol is a β-AR antagonist that decreases morbidity and mortality in patients in heart failure and post acute myocardial ischemia and is said to promote, relative to other β-antagonists, a survival advantage to patients (15), although this result is controversial (16). Carvedilol also has α1-adrenergic blocking activity that contributes to its therapeutic effect, and a number of other actions that have been suggested as contributing to its survival advantage. A recent study suggests that functional selectivity may also contribute to this advantage.
Carvedilol has a unique profile of action among β-blockers in that it antagonizes Gs-mediated signaling while stimulating β-arrestin–mediated signaling (17). Animal studies have shown that β-arrestin signaling may exert a cardioprotective effect, while Gs-dependent AC activation is cardiotoxic (18). If this is the case in human heart failure, this unique profile of action may contribute to the superior effectiveness of carvedilol. This mechanism could also provide the basis for developing new β-antagonists based on their functional selectivity profile.
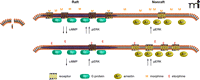
Functional selectivity of μ-opiate receptor ligands and membrane raft domains. Upon binding of morphine (top panel, left), the receptor conformation has a greater affinity for Gi, which is localized to the raft domain. Signaling through Gi inhibits adenylyl cyclase to decrease cyclic AMP and leads to increased phosporylation of ERK. The morphine-activated receptor has a lower affinity for β-arrestin––localized to nonraft domains (top panel, right)––which leads to ERK phosphorylation. The etorphine-bound receptor (bottom panel, right), favors coupling to β-arrestin, leading to greater ERK activation and relatively lesser inhibition of adenylyl cyclase. Arr, β-arrestin; cAMP, cyclic adenosine 3′,5′-monophosphate; E, etorphine; ERK, extracellular signal–regulated protein kinase; Gi, inhibitory G protein; M, morphine.
Acknowledgments
Thanks to Seksiri Arttamangkul, Ren Ostrom and John Williams for helpful comments.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Mark A. Simmons, PhD, is a Professor of Pharmacology at the Northeastern Ohio Universities College of Medicine. His research interests include the pharmacology of tachykinin receptors. E-mail simmons{at}neoucom.edu; fax 330-325-5912.

