Caenorhabditis elegans in Parkinson’s Disease Drug Discovery: Addressing an Unmet Medical Need
Abstract
It has been over forty years since dopamine neuron degeneration in the substantia nigra and Lewy body formation within surviving cells were described as the pathological hallmarks of Parkinson’s disease (PD). Although research in the intervening decades particularly in the last twenty-five years has yielded a variety of robust animal models and invaluable mechanistic insights into PD-associated neuronal dysfunction and cell death, therapeutic agents have not been forthcoming to alter the course of PD. Recently, the screening of experimental therapeutics for PD has been pursued through the use of genetically tractable models, such as the nematode Caenorhabditis elegans. This simple worm remarkably recapitulates the basic cellular and molecular pathways associated with PD, is amenable to facile genetic methods, and through the use of high-throughput screening technologies, provides powerful new opportunities for the in vivo identification of therapeutic targets. In this review we briefly describe the utility that the C. elegans model system may have for PD drug discovery.

Introduction
The physical manifestations of Parkinson’s disease (PD) were first detailed by the British neurologist James Parkinson, who in 1817 published his observations of affected London pedestrians in An Essay on the Shaking Palsy (1). PD is now recognized as the second most prevalent neurodegenerative disease, affecting ten percent of individuals age sixty-five or older, or more than four million people worldwide. PD is a chronic and progressive disorder, presenting not only the cardinal symptoms of tremors, bradykinesia (slowness of movement), and rigidity, but also a host of other and increasingly recognized non-motor dysfunctions, including orthostatic intolerance, constipation, bradyphrenia, and depression (2–4). Individuals with PD often survive two decades or more following the appearance of symptoms, with a mortality rate fifty percent higher than that of similarly aged individuals. Patients generally do not die directly from PD, but rather from unrelated illness or complications associated with the decrease in mobility, such as pneumonia and physical injuries from falling (3). More than a century and a half after Parkinson described physical symptoms of the disease in his essay, the loss of dopamine neurons (greater than eighty percent in the pars compacta of the substantia negra) and the formation of protein aggregates, termed Lewy bodies, or Lewy neuritis, were recognized as pathological hallmarks of PD (3, 5). Neuropathological effects, also apparent in the locus ceruleus, raphe nucleus, and nucleus basalis, are accompanied by abnormal serotonin, norepinephrine, and choline levels (6). Alterations in these neurotransmitter systems, as well as in limbic structures and neorcortex, likely contribute to PD-associated non-motor deficits (7).
Etiological and pathological evidence suggests that there are both genetic and environmental components to the development of the more commonplace sporadic forms of PD, or idiopathic PD. In the less common, familial form of PD, at least five genes have been implicated (8). Although familial forms account for about ten percent of all PD cases, the biochemical and cellular similarities in the pathogenesis of idiopathic and familial PD suggest that both may involve similar molecular mechanisms of cellular dysfunction and death, including oxidative stress, protein aggregation, and proteosomal or mitochondrial dysfunction. The identification of mutations that are linked to PD has significantly aided the development of genetic model systems for investigations into the molecular basis of PD.
Epidemiological evidence suggests that exposure to environmental toxins may also play a role in PD. Exposures to pesticides and insecticides—in particular, paraquat, maneb, and rotenone—have been associated with an increased incidence of the disease (9–14). These compounds are toxic to dopamine neurons in animal models of PD and inhibit mitochondrial complex I, consistent with the hypothesis that mitochondrial dysfunction may contribute to the disease. Exposures to a number of heavy metals, particularly manganese, have also been linked with the development of parkinsonism (15–17). The contribution of manganese to the increased incidence of disease among welders is currently the subject of litigation in the US courts.
Pharmacological treatment of PD, including the use of compounds that affect dopamine-associated systems, are primarily limited to the level of symptomatic control. Levodopa (l-DOPA), an amino acid common to plants and animals, has been the primary and most efficacious medication used, for nearly forty years, to treat PD (4). Largely prescribed to treat motor symptoms, l-DOPA is a precursor of dopamine and is sequestered in synaptic vesicles that function in dopamine neurotransmission. Pharmaceutical l-DOPA is often formulated (e.g., Sinemet or Madopar) with a decarboxylase inhibitor to prevent peripheral turnover of the prod-rug into dopamine and thereby mitigate side effects (i.e., nausea and gastrointestinal disruption). Notably, within five years after initiating l-DOPA treatment, over fifty percent of patients develop motor fluctuations and dyskinesias that can be as disabling as the primary symptoms (3). The molecular basis of these side effects is not clear but may reflect either the saturation of dopamine-sequestering mechanisms, as neuronal survival rates diminish, or downstream modification of postsynaptic neuronal receptors. Therapeutic regimens may also include D1 and D2 receptor agonists (e.g., apomorphine, bromocriptine, pramipexole, and ropinirole), monoamine oxidase inhibitors (e.g., selegiline and rasagaline), and anti-cholinergic agents (e.g., trihexyphenidyl, benzotropine, and amantadine) (5). Amantadine, first identified as an anti-influenza therapeutic, may alleviate PD symptoms by increasing synaptic dopamine release, inhibiting dopamine reuptake into the presynaptic terminal, increasing D2 receptor expression levels, or blocking NMDA receptors (3, 6). Amantadine also has antidyskinetic properties, but as is true for all the drugs mentioned above, its efficacy lessens over time, with an increase in both physical and mental adverse effects.
Currently, there is no cure for PD and no therapeutics are available that can slow the progressive loss of dopamine neurons or prevent Lewy body formation. Why have disease-modifying drugs been so difficult to identify for PD? A major issue has been our limited knowledge of the molecular mechanism(s) that underlie the pathogenesis of PD, which has precluded the identification of targets of intervention. The recent identification of pathogenic mutations in at least five genes has fueled unprecedented investigation into the pathobiology of PD, which is bound to yield fruits in the foreseeable future (7).
The use of invertebrate models such as the nematode Caenorhabditis elegans offers opportunities to take advantage of human molecular genetic studies by incorporating facile genetic and chemical screens. In this review, we briefly describe how this animal model may facilitate the identification of genes and molecular pathways involved in PD, information that is crucial to the development of disease-modifying therapeutic targets and chemical leads.
Human physiology in a Worm
Less than a decade ago, C. elegans became the first multi-cellular eukaryotic genome to be successfully sequenced and is the centerpiece for two Nobel Prizes. The 2002 Award to Sydney Brenner, Robert Horvitz, and Johns Sulston highlighted the strong genetic and molecular similarities in organ development and programmed cell death between the nematode and humans, and the 2006 Prize to Andrew Fire and Craig Mello further emphasized this conservation with the identification of fundamental mechanisms of gene regulation. These advances provided valuable insight into cellular physiology and neurodevelopment, paving the way for the use C. elegans to elucidate molecular mechanisms involved in human disease.
C. elegans contains approximately 20,000 genes, virtually identical to the number of genes determined in humans (18, 19, 20). Genes and molecular pathways involved in cell and organism development, apoptosis, and neurotransmission [e.g., dopamine, serotonin, acetylcholine, glutamate, and γ-aminbutyric acid (GABA) systems] are highly conserved (Figure 1⇓) (21–24). Accordingly, C. elegans models have been established for a host of diverse human disorders, ranging from Parkinson’s, Alzheimer’s, and Huntington’s diseases to cancer and immunological dysfunctions (25–27). Furthermore, neuroreactive compounds, including neurotransmitter agonists and antagonists, anesthetics, and immunosupressants, that have been widely applied to humans, interact with the orthologous worm proteins, replicating the biochemical and behavioral phenotypes typical of vertebrate species (28, 29).
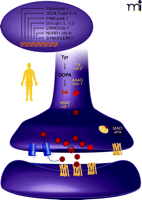
Conservation of Parkinson’s disease–associated genes inC. elegans. Genes associated with PD are listed in the oval along with orthologs (italics) in C. elegans. (As indicated by the question mark, α-synuclein represents the only PD-associated gene that does not have a strong ortholog.) A dopamine neuron schematizes known and predicted genes involved in dopamine biosynthesis and metabolism Red circles represent dopamine; blue cylinders represent dopamine transporter; heptahelical structures represent pre- and postsynaptic dopamine receptors. Protein locations are putative. AAAD, aromatic L-amino acid decarboxylase; MAO, monoamine oxidase; TH, tyrosine hydroxylase; VMAT [the activity of which is indicated by appearance of black circles (membranes of dopamine-containing synaptic vesicles)], vesicular monoamine transporter.
If a primary onus of simple genetic model systems is to provide significant experimental advantages over the use of mammalian systems, the C. elegans meets or exceeds demands, especially in terms of facilitating genetic approaches (see www.wormbook.org). The worm’s diminutive size (about 1 mm in length), large number of offspring (about 350), short generation time (3.5 days), and ease of growth in the laboratory (on bacterial lawns and in multiwell microtitre plates), allow for rapid and inexpensive production of animals (30, 31). The animal is anatomically simple, containing about 1000 cells, with a third of these differentiated as neurons. Notably, the nervous system has been fully mapped at the ultrastructural level, permitting the identification of chemical and electrical synapses in the whole organism (32). Primary cultures, first developed by Laird Bloom in the early 1990s, allow for cell-specific and electrophysiological studies, as well as the generation of tissue-specific cDNA libraries (33–36). Worm strains can also be stored for many decades at −80° C or in liquid nitrogen.
C. elegans is amenable to forward genetics, allowing the assignment of genes—without prior knowledge of their function—to a particular cellular process or phenotype, in as little time as a week (30, 37, 38). The nematode is also easily crossed, allowing for the ready generation (within days) of strains with particular genetic mutations. Transgenic animals are also readily engineered, and worms that express the green fluorescent protein have permitted the in vivo visualization of cellular and metabolic processes. The world-wide C. elegans community currently shares over 8000 mutant strains that are freely available from the C. elegans Genetic Stock Center (see http://elegans.swmed.edu). C. elegans is also amendable to reverse genetic approaches that allow for the knockdown of gene products in vivo. A particularly powerful and rapid method of reducing gene expression is afforded by feeding the animals E.coli that express dsRNA (39–41). Bacterial feeding libraries, containing roughly 20,000 individual E. coli strains, each expressing dsRNA to a specific C. elegans gene, allow for knockdown of most of the known genes in the C. elegans genome, allowing for facile phenotypic analysis in whole-genome screens (42).
C.elegans: A Model Dopamine Neuronal System for PD
Biochemistry and Neurotoxins
On the cellular and molecular level, the dopamine neurons of C. elegans recapitulate the characteristics of mammalian dopamine neurons. But unlike vertebrates, which contain tens of thousands of dopamine neurons in the mid brain, the hermaphroditic C. elegans organism contains only eight dopamine neurons, thereby simplifying a wide array of investigations (Figure 2⇓) (26, 43). These eight dopamine neurons, believed to be mechanosensory neurons, are involved in basal motor activity, habituation, egg-laying, and defecation. They display all the mammalian enzyme activities involved in dopamine biosynthesis, storage, transport, and signaling, including GTP cyclohydrolase I, tyrosine hydroxylase, aromatic amino acid decarboxylase, and vesicular monoamine transferase. They also recapitulate mammalian dopamine receptors and the dopamine transporter (DAT), which is the target of drugs of abuse and mediates the PD-associated activity of certain neurotoxins (see Figure 1⇑) (24, 44). Importantly, C. elegans also contains enzymes responsible for dopamine catabolism, including monoamine oxidase (MAO) and catechol-O-methyl transferase (COMT); consequently, the organism produces the dopamine catabolites 3,4-dihydroxy-phenylacetic acid, 3-methoxytyramine, and homo-vanillic acid (24, 45).
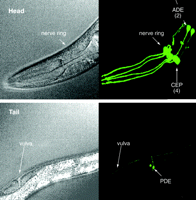
Dopaminergic neuroanatomy in theC. elegans hermaphrodite. Adult wild-type worms contain eight dopamine neurons, which can be visualized in transgenic animals that express the green fluorescent protein (see text for details of dopaminergic neuron–specific expression of GFP). The head of the animal (upper panel; the pharynx is labeled for visual orientation) includes two anterior deirid (ADE) neurons and four cephalic sensilla (CEP) neurons; arrows point to cell bodies. The tail of the animal (lower panel; the vulva is labeled) includes two posterior deirid (PDE) neurons; arrows point to cell bodies. Live animals were imaged through differential interference contrast microscopy (left images) and confocal epifluorescence microscopy (right images). Reproduced with permission from (26).
Currently available vertebrate models rely on either familial PD–associated genetic manipulations or on toxin exposure to reproduce the molecular basis of dopamine neuronal death and associated pathologies. Both types of models have also been successfully employed in the worm. Moreover, transgenic worms that express the green fluorescent protein (driven by the DAT promoter) provided the ground-breaking opportunity to visualize an entire population of dopamine neurons within a living animal (44). Indeed, novel in vivo investigations of dopamine neuronal death became possible by exposing these worms to the same neurotoxins and mitochondrial complex I inhibitors (e.g., 6-OHDA and MPP+) that had been used to model PD symptoms in vertebrates (26, 45); worms that were treated with a DAT antagonist or that were deficient in the DAT-encoding gene were accordingly protected against neuronal loss. The pesticide rotenone, another complex I inhibitor used to model PD in vertebrates, adversely affects worm mortality and dopamine neuron vulnerability, likely by blocking mitochondrial respiration and thereby recapitulating mammalian models (46). We have also found that several transition and heavy metals, including copper, iron and manganese, can confer dopamine neuronal cell death in C. elegans (47, 48). As in mammalian systems, manganese sensitivity is partially dependent on the C. elegans ortholog of DMT-1, the human divalent metal transporter, and is associated with adverse effects upon the mitochondria complex I and membrane potential (47–49).
Genetics
Familial PD, which accounts for less than ten percent of all known cases of PD, has been associated with the genes that encode α-synuclein, parkin, DJ-1, PINK-1, and LRRK2 (Figure 1⇑). Mutations in α-synuclein formed the first evidence of a genetic basis for familial PD, and although its function is largely unknown, α-synuclein may play a role in synaptic vesicle docking, dopamine biosynthesis, DAT function, and neuroprotection (50–54). Although α-synuclein represents the only PD-associated gene that does not have a strong ortholog in the worm, overexpression of the human protein recapitulates some of the biochemical, behavioral, and pathological characteristics of the human disease. Overexpression of either the wild-type or a familial PD–linked mutant form of human α-synuclein in the worm causes dopamine neuronal death, motor deficits, aggregate formation, disruption of vesicle docking, and as is the case in vertebrates, protects against some forms of oxidative stress (55, 56) (Figure 3⇓). Notably, C. elegans microarray studies indicate that α-synuclein causes significant changes in molecular pathways involved in proteosomal and mitochondrial function (57).
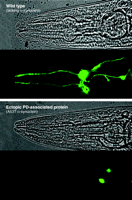
Degeneration of Dopamine neurons in response to a PD-associated protein. Ectopic expression of the human α-synuclein A53T isoform causes C. elegans Dopamine neurons to degenerate. The anterior (see Head; Figure 2) of the adult wild-type worm (upper panel) shows normal compliment of Dopamine neurons, whereas the A53T α-synuclein–expressing animal (lower panel) shows loss of cell bodies and processes. Only four of the six neurons are clearly visible in the wild-type worm because of the focal plane. Only two neuronal cell bodies (the ADE pair; see Figure 2) and part of the processes are present. Reproduced with permission from (58).
Manipulation of PD-associated genes, including knockdown and deletion, evoke phenotypes of C. elegans that are consistent with vertebrate studies. Mutations in parkin, which is an E3 ubiquitin ligase that targets proteins for proteosomal degradation, result in increased sensitivity to 6-OHDA, confer hypersensitivity to stressors of the endoplasmic reticulum, and cause cellular degradation (46, 58–60). Expression of the familial PD–associated mutant form of α-synuclein is lethal to worms that lack parkin, suggestive of increased cellular stress. Genetic knockdown of a worm ortholog of DJ-1 leads to increased sensitivity to rotenone (45). On the other hand, the mammalian wild-type LRRK2 protein, a multidomain protein of largely unknown function, protects worms against rote-none toxicity, whereas a PD-linked mutant form of the protein does not (61). These studies collectively underscore the utility of C. elegans as a model of molecular pathways associated with PD.
Elaboration of PD-Associated Pathways
It is clear that C. elegans recapitulates critical physiological components of PD and vertebrate models, but how can this system identify novel PD-associated therapeutic targets? As already mentioned, simple forward genetic screens can be effected on the order of days to weeks, and such screens might be used to identify modifier proteins involved in PD phenotypes. For example, transgenic worms that express the green fluorescent protein within dopamine neurons (see above) provide the basis for genetic screening. Specifically, the dopamine neurons in the progeny of mutagen-treated self-fertilizing animals could be examined for resistance to the PD-associated effects of 6-OHDA. Because DAT is required for the entry of 6-OHDA into the cell, viable fluorescent progeny would likely have mutations within proteins related to DAT functionality or involved in molecular pathways (e.g., cell death pathways) that affect the sustainability of dopamine neurons. Proteins identified in this approach are potential candidate targets for neuroprotective therapeutics. Importantly, this screen is performed in vivo, so that the complete neurochemical and cellular pathways of the whole organism are functional and intact, which significantly increases the likelihood of identifying potential therapeutic targets. Our initial screen has identified a number of mutants that have varying resistance to 6-OHDA. As expected, several of the mutations are found within DAT, providing proof-of-concept that proteins involved in PD-associated toxin-induced cell death can be identified (62).
Reverse genetic screens, as introduced above, are also attractive. RNAi bacterial feeding libraries provided to worms cultured in 96-well plates can rapidly and inexpensively facilitate the genome-wide identification of proteins involved in neurodegeneration or PD-associated pathologies (29, 63). Recently, Nollen and colleagues screened over 16,000 bacterial clones, representing almost 85% of all C. elegans genes, and identified 80 genes that are involved in human α-synuclein aggregation (64). Thus, even though a protein may not be evolutionarily conserved (e.g., α-synuclein), it can nevertheless be assessed in this model system to identify modifiers of PD. In this instance, the proteins identified included those involved in vesicle transport and herbicide detoxification, aging, and homologs identified from other PD models (64). Other researchers have utilized RNAi methodology to identify putative modifiers of α-synuclein toxicity and genes involved in protein trafficking and transport (65).
The ease of genetic manipulation in C. elegans also offers an approach to assess candidate genes identified from DNA microarray or proteomic studies on postmortem patients. RNAi-mediated knockdown of the expression of the orthologous proteins in the worm and its effect on dopamine neuron vulnerability or other aspects of PD is one means of exploiting the high genetic and molecular conservation between worms and vertebrates. Functional genomic approaches based on C. elegans should facilitate investigation into the roles particular genes may play in PD pathologies and thus the identification of much-needed drug targets (66, 67).
Identification of Novel Drug Leads and Mode of Action
In addition to the rapid identification of mutants, facile engineering of transgenic animals, and targeted knockdown of protein expression, the rapid development of transparent worms in multi-well plates accommodates rapid compound screening and phenotypic analysis of neurons (63). C. elegans are permeable to aqueous and organic molecules, although the tough, outer cuticle may necessitate high concentrations. Once compounds are absorbed, they may readily diffuse into the worm’s nervous system, as the animals lack a functional blood-brain barrier. Fortunately, C. elegans grows well in 1–2% DMSO, a common solvent and anti-oxidant in high-throughput drug libraries (28, 68).
To date there has not been a large-scale chemical screen instituted in C. elegans to identify compounds that could ameliorate PD-associated phenotypes, but worm-based compound screening has identified drug leads for diabetes and arrhythmias, and smaller-scale screens have identified compounds that protect dopamine neurons from cell death. Thus, the utility of the C. elegans model for high-throughput screens, using simple fluorescent plate readers and compound scopes, has been established (29). In a screen partially supported by the Michael J. Fox Foundation for Parkinson’s Research, over 2000 compounds were screened for their ability to inhibit the 6-OHDA–induced death of dopamine neurons (68, 69) (Figure 4A⇓). The screen yielded a number of neuroprotective compounds, including the NMDA receptor antagonist dextromethorphan, the antimicrobrial agent colistimethate, the dopamine D2 receptor agonists quinpirole and bromocriptine, and the mixed D1/D2 agonist dihydrexidine. Importantly, DAT antagonists that would inhibit 6-OHDA uptake were also identified (69–71). Intriguingly, dextromethorphan protects against MPTP toxicity in rodent models, and D2 agonists have been previously implicated as neuroprotective in 6-OHDA–lesioned rats, suggesting that this assay may be useful to identify and reposition drugs or novel leads for application to PD (see below), depending on the chemical library screened (72, 73).
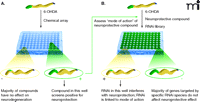
In vivo assays of dopamine neuron degeneration in microtiter plates: Identification of therapeutic leads and modes of action. Transgenic worms in which the green fluorescent protein identifies functional dopamine neurons allow for the screening of exogenous compounds (e.g., chemical libraries) and endogenous molecules (e.g., RNAi libraries) potentially relevant to Parkinson’s disease. A. The effects of compounds upon dopamine neuron degeneration (here, induced by 6-OHDA) can be observed as a function of GFP fluorescence. Several hundred transgenic embryos or L1 larvae are placed in wells in a microtiter plate, with each well containing a potential therapeutic lead. After the appropriate incubation time (e.g., four days), each well that emits a strong fluorescent signal, suggesting a neuroprotective effect, will be selected for further investigation. B. A selected compound’s mode of action can be assessed through the use of an RNAi library. Young transgenic worms are incubated in wells, with each well containing a bacterial clone that expresses a dsRNA that will target a specific C. elegans gene. Wells in which animals show little or no fluorescence likely represent the reduced expression of a protein that is a necessary for the efficacy of the given compound. Modified and reproduced with permission from (69).
One strategy for identifying novel compounds that could be relatively quickly developed into drugs is to construct chemical libraries that allow movement from the primary hit stage in C. elegans to potential therapeutic leads that can be validated in mammalian systems. In the pharmaceutical industry, many bioactive molecules have been pursued through synthetic processes to optimize drug-like properties associated with absorption, metabolism, and disposition—only to fail to show efficacy thereafter. These failed attempts represent diverse chemical libraries, enriched in molecules partly characterized in rodents, which might offer new opportunities for drug discovery using C. elegans.
C. elegans drug screening paradigms (Figure 4B⇑) also provide in vivo opportunities to identify the mode of action of novel leads, whether these originate from C. elegans or other screening sources. Mode-of-action and target validation may not be adequately addressed in tissue culture or vertebrate models that, owing to expense and labor, significantly extend the time for drugs to reach the marketplace. The strong conservation of molecular pathways between human and worm, combined with C. elegans utility for high-throughput screening in an intact animal, has the potential to identify the molecular targets and pathways of potential drugs more quickly than would be possible in rodent models. The combination of facile technologies in intact organisms is uniquely permits targets and pathways to be explored for therapeutic relevance in their native biological environments (74). Meaningful mode-of-action studies in C. elegans have been accomplished for human anticancer, anticonvulsant, and antidepressant compounds (29).
The incorporation of C. elegans into mode-of-action studies also has potential in the early identification of toxicity, another significant barrier in moving drugs to the market (75–77). Putative therapeutics may pass early efficacy and mid stage surrogate toxicology tests in drug development, only to be discarded later when adverse events arise unexpectedly. Complex drug interactions may be better predicted in mode-of-action studies that are performed in whole organisms, such as the facile C. elegans assays discussed above. Phenotypic readouts such as organism viability or feeding behavior are exclusive to such assays, but subtle abnormalities of biochemistry, only evident in physiologically relevant environments, can also be detected (78). For example, we were able to identify phospholipidosis-producing compounds that had gone undetected during in silico screening and otherwise raised suspicions only months later in rodent testing (79). Admittedly, toxicity mechanisms are often species specific, but the utility of C. elegans models lies in the rapid opportunity to assess adverse reactions in a highly conserved system. Although these models may inevitably miss some toxicities—as do all models—the identification of even a relatively small number of problematic leads will greatly aid drug discovery efforts.
Conclusions
New technologies are consistently emerging, promising to transform drug discovery through the use of C. elegans models. The Complex Object Parametric Analyzer and Sorter (COPAS; Union Biometrica, MA) is essentially a fluorescent activated worm sorter that can rapidly dispense (greater than one worm per second) specific numbers, sizes, or fluorescent intensities of living worms for high-throughput screening of neuroprotective compounds. Neuroprotective phenotypes could quickly be examined in secondary assays, and suppressor chemical screens (described above) could quickly be incorporated to identify relevant genes. Earlier studies using the sorter have successfully screened for genes involved in a number of biological processes related to aging, innate immunity, gene regulation, and toxicological responses (80–82).
Other more recent developments to identify compounds that may inhibit PD-associated phenotypes include an on-chip high-throughput small-animal technology (83). Hundreds of live worms can be immobilized in individual chambers containing lead compounds on a microfluidic chip, so that images can be generated with subcellular resolution. Animals that have specific subcellular phenotypes, for example those that lack α-synuclein–containing aggregates, can be rapidly sorted, thereby identifying compounds for further analysis. C. elegans is poised to make significant contributions to PD drug target identification and validation and the development of novel therapeutic compounds. The organism’s high degree of genetic and cellular similarity to humans, its utility for whole-animal high-throughput genetic and chemical screens, and its amenability to genetic manipulation will likely make this small nematode a powerful ally in determining the molecular pathways involved in PD and in the development of novel therapeutic agents.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Timothy Ryan, PhD, is a Senior Research Advisor in the Department of Integrative Biology at Lilly Research Laboratories. He earned his doctorate in Molecular Biology and Toxicology from the Biotechnology Center at Utah State University. His postdoctoral research, at Pharmacia & Upjohn in the Department of Investigative Toxicology, focused on the role of oxidative stress and antioxidant therapies in toxicity and disease. At Lilly Research Laboratories, his laboratories utilize functional genomics to identify and validate new drug targets, biomarkers, and mechanisms of pharmacology/toxicity in the early phases of drug development.

Kalpana M. Merchant, PhD, is Chief Scientific Leader for Integrative Biology at Lilly Research Laboratories in Indianapolis. She received her doctorate in neuropharmacology from the University of Utah and worked in academia before establishing her career in CNS drug discovery before joining Lilly. She specializes in translational approaches for the discovery and development of drugs for neuropsychiatric and neurodegenerative disorders. Under her leadership, the Integrative Biology group is accountable for target and biomarker discovery and validation and providing reagent support for research initiatives in translational medicine and toxicology. Her own laboratory focuses on approaches to Parkinson’s disease. She serves on the Scientific Advisory Board for Michael J. Fox Foundation for Parkinson’s Research, NIH Study Sections, and contributes to several national and international professional societies.

Richard Nass, PhD, earned his doctorate at The Johns Hopkins University School of Medicine and pursued postdoctoral work at Vanderbilt University, where he developed the first C. elegans models for Parkinson’s disease. He is currently an Associate Professor of Pharmacology and Toxicology at Indiana University School of Medicine, which also includes primary appointments in the Stark Neuroscience Research Institute and the Center for Environmental Health. His laboratory interests are to identify and characterize the molecular components involved in dopamine neurodegeneration and Parkinson’s disease, and to screen for pharmaceuticals and genetic pathways that protect against cellular death. He also explores the molecular basis of heavy metal–induced neuropathologies and neurodevelopmental defects. He is primary editor of the recently published Parkinson’s Disease: Molecular and Therapeutic Insights from Model Systems (Elsevier). Send correspondence to RN. E-mail ricnass{at}iupui.edu; fax 317-274-7868.

