Short and Sweet: Evolution of a Small Glycopeptide from a Bladder Disorder to an Anticancer Lead
Anticancer drug discovery paradigms have evolved rapidly in concert with advances in scientific knowledge and technological breakthroughs. Although protein biologics like monoclonal antibodies are being developed and approved, small-molecule inhibitors still hold the spotlight as highly anticipated clinical therapies; hence, most modern drug discovery efforts remain focused on low molecular-weight modulators of “target” processes that contribute to the pathological state of the disease (1). When a valid protein target is identified, libraries of compounds are often screened in high-throughput fashion to find leads, which are further optimized and cycled through other assays and into animal models, culminating, if fortunate, in preclinical development. In some cases, the disease itself becomes the source of agents that are directly linked to its pathology, and the function of the agent is the critical clue in how to combat the ailment. Such is the case for a novel, endogenous small molecule that causes many of the problems associated with a disease called interstitial cystitis (IC) [also termed painful bladder syndrome (PBS)] (2).
IC/PBS (3) is a condition of the bladder marked by thinning, ulceration, or both, of the bladder epithelial lining, causing pain and discomfort that is often severe. The pain is thought to be a consequence of exposure of sensory nerve cells to urinary components arising from the epithelial necroses. Patients are much more likely to be women than men (~8:1), and IC/PBS is often co-morbid with other inflammatory diseases, such as irritable bowel syndrome. There is no known cause of IC/PBS, but since the late 1990s, groundbreaking work has led to the identification of an agent produced in the bladders of IC/PBS patients that may elucidate the origin and treatment of the disease. In 2004, the structure of a factor from the urine of IC/PBS patients that strongly inhibited normal bladder epithelial cell growth was published (4); this factor seemingly was the cause of many symptoms of IC/PBS. The molecule, called antiproliferative factor (APF), is a nine-residue sialolglycopeptide with sequence identity to a motif in the sixth transmembrane region of human Frizzled-8 protein (Frz8), a Wnt ligand receptor. The glycan (i.e., the oligosaccharide portion) proved to be a trisaccharide, Neu5Acα2-3Galβ1-3GalNAc, alpha-O-linked to a threonine residue at the N terminus of the peptide (Figure 1⇓). APF inhibits normal bladder cell proliferation at sub-nanomolar concentrations, offering evidence to suggest that APF causes the bladder epithelial thinning seen in nearly all IC/PBS patients. APF also caused a dramatic reduction in the amount of heparin-binding epidermal growth factor-like growth factor (HB-EGF) in urine (5), and caused attenuation of tight-junction formation through the reduced expression of several proteins involved in this process (6).
Although these discoveries are a boon to IC/PBS biology, what does this have to do with cancer? APF is a powerful negative growth factor, and it was surmised that if it can shut down the growth of normal bladder cells, it is possible that it may function against bladder tumor cells with equal or greater potency. In the original structural studies (4), APF markedly inhibited the proliferation of T24 bladder carcinoma cells. Subsequent studies showed that APF can also inhibit kidney cancer cell growth (7) and that the growth inhibition of normal bladder and T24 bladder carcinomas was p53 dependent (8). Aggressive efforts are ongoing to identify other tumor cell lines that are sensitive to APF treatment and to elucidate its specific mechanisms of action.
Although all these results are very exciting and provocative, the medicinal chemistry of APF is wrought with much more puzzle than promise. For example, although the saccharide portion of the molecule is an absolute requirement for biological activity, such activity does not depend on the presence of sialic acid: both APF and asialo-APF (as-APF) are essentially equipotent (4). The hexose-hexosamine disaccharide was originally hypothesized to be a lactosamine (Galβ1-4GlcNAc) unit; although this proved incorrect, a synthetic APF with alpha-O-linked lactosamine to the N-terminal threonine was equally as potent as as-APF. The alpha-O-link to the threonine residue is an absolute requirement, because saccharides linked in the beta-configuration are inactive. These findings may have consequences as to the identity of specific cellular receptors for APF (vide infra).
We recently completed a structure-activity relationship (SAR) study of the peptide portion of APF (9), finding that very minor changes to the peptide sequence either greatly attenuated or completely abrogated activity, as evaluated by inhibiting 3H-thymidine incorporation of normal bladder cells (Box 1). One very intriguing analog replaced l-proline with its d-enantiomer. This derivative was completely inactive in inhibiting bladder cell proliferation but it blocked natural APF activity and reversed some of the consequences of APF function (10). Hence, treatment of IC cells with d-proline–APF significantly increased cell proliferation, increased zonula occludens-1 and claudin 1, 4, and 8 expression, and decreased paracellular permeability of these cells, all consequences of normal APF treatment (6). Thus, d-proline–APF may turn into an excellent lead candidate for therapy in IC/PBS patients.
Structure-Activity Relationship (SAR) Analysis of APF
Acetylation of the N-terminal amine of APF, a carboxyamide group at the C-terminus or extension of the as-APF sequence on either termini by one additional amino acid resulted in an approximate two orders of magnitude drop in potency. Replacement of the N-terminal threonine with an appropriately glycosylated serine reduced potency by four orders of magnitude, whereas keeping the substituted serine and replacing the adjacent valine with a leucine recovers, by two orders of magnitude, the attenuated potency. If the original threonine is maintained and Val2 is replaced with a leucine or tyrosine, the activity disappears completely. The only changes that can be made that maintain full potency are the removal of the C-terminal alanine (to give an 8-mer), and replacement of the Pro4 with pipecolic acid (Pip), a six-membered ring piperidine analog of the five-membered pyrrolidine ring of proline. These two are the only fully potent analogs out of nearly forty tested. Alanines located in position 5 and 9 were found to be optimal for antiproliferative activity. Ala9 can be removed without loss of activity, but any substitution leads to a significant decrease in potency or to total inactivation. A substitution of Val6–8 with less hydrophobic amino acids has detrimental effect on the activity. When they are replaced with alanines the activity drops to 1%, whereas substitution with glycines leads to even less potent compound (0.01%). Moreover when even a single Val from the segment was substituted with putative “helix-disrupting” amino acids, the resulting compound was inactive. A summary of the APF SAR is illustrated in Figure 2⇓.
There is little doubt that the function and potential therapeutic value of APF either as a tool or as a model for future commercial agents shall garner keen interest from both the urologic and cancer research communities. But with regard to only the basic science of APF, perhaps the most thought-provoking aspect of the APF discovery and evaluation is its biosynthesis: specifically, the glycobiology of APF. APF is a very small, secreted, negative growth factor glycopeptide with virtually no precedent in the medical literature. There have been a smattering of reports of other “glycopeptides” with antiproliferative activity, but they were all of small protein size (> 10 Kd) (11) and none have been characterized beyond aminoacid sequence data or pro-protein origin (i.e., proteolysis products of endogenous blood proteins). APF, on the other hand, contains only nine residues, all of them hydrophobic except for the terminal glycosylated threonine. The sequence of APF is identical to a putative transmembrane domain of an important protein involved in mitogenic signaling, Frz8. Hence the first paradox: Why would a transmembrane domain motif be glycosylated? If APF is truly from a transmembrane segment––indeed, it is unknown whether APF arises from specific proteolytic cleavage of Frz8 or whether APF interrupts Frizzled signaling––how was Frz8 processed to yield this secreted small molecule? Because the sialic acid is not required for activity, and the majority of the analog studies were performed with as-APF, is it possible that natural APF is simply a prodrug of as-APF via the action of an in vivo neuraminidase? And what about the Galβ1-3GalNAc disaccharide? This disaccharide, also known to carbohydrate scientists as the Thomsen Friedenreich (TF) disaccharide, is a tumor-associated carbohydrate antigen used as an epitope in tumor vaccine development (12). Why is it covalently attached to the nonapeptide sequence? Most complex vertebrate O-glycan synthesis begins with the transfer of GalNAc in the alpha configuration to acceptor serine or threonine residues via a UDP-GalNAc donor; the subsequent beta-1-3 transfer of Gal yields the TF disaccharide, which is labeled as the core-1, O-linked structure. In normal tissue, further elongation leads to larger and more branched glycans and, in many cases, these are terminated by sialic acid (13). Addition of the alpha-2-3 sialic acid in the case of APF evidently serves to terminate further elongation of the trisaccharide by other sugar transferases. If the sialic acid is not necessary for activity in vivo, could the sugar be the critical portion of the pharmacophore that mediates the activity of APF? We had hypothesized that because the peptide portion is extremely hydrophobic and the sugar “protrudes” from the N terminus, the APF peptide may interact with the membrane and hence present the sugar portion of the molecule extracellularly for recognition (e.g., by its receptor, etc.), akin to glycolipid patching in cell membranes. Recent results have shown that cytoskeletal-associated protein 4 (CKAP4, also termed p63) is a receptor for APF in IC cells and that “knockdown” (i.e., reduced expression via short interfering RNA) of this protein reduces the APF-mediated effect on proliferation (14). Not much is known about the ligands for CKAP4/p63, its three-dimensional structure, its role in mitogenic signaling, or whether sugar moieties on CKAP4 ligands are important for binding. These critical questions remain to be answered to further our understanding of the molecular mechanisms of APF function in IC/PBS pathobiology.
In the cancer arena, it is entirely possible that APF function may be mediated by other recognition partners or cellular events. If the aforementioned “glycolipid”-like theory is valid, presentation of TF or sialo-TF saccharides in clusters at the membrane surface may engage lectins (or possibly galectins––which are lectins that specifically bind to beta-galactoside moieties on glycopeptides or glycoproteins––that may bind both TF antigen and lactosamine, vide supra) in a multivalent fashion to modulate signaling through carbohydrate-protein binding or carbohydrate-carbohydrate interactions (15) between adjacent cells. From our SAR studies and the observed extraordinary lability of APF activity to minor structural changes, it seems likely that APF may require a physical state––other than as a simple monomer––to elicit its function. NMR studies from our laboratory (unpublished) have shown that APF does not adopt a specific structure in water and buffer solution, which is not surprising for a peptide this size. Some structure in the C terminus is obtained when high concentrations of trifluoroethanol are added, but nothing to suggest a large degree of defined secondary structure or aggregation in solution. The AXXXA motif, often found in the C termini of proteins, is involved in helix-nucleation in proteins (16). Under the proper circumstances, the peptide portion of APF may form a defined structure, allowing APF to interact specifically with cellular receptors and potently inhibit proliferation. This, too, is only conjecture, as there is no structural data available of APF bound with its receptor. The biophysical characterization of APF in various milieus and with putative receptors is also an active area of research.
Along with the possibility of deriving a therapeutic based on APF structure and function, could the discovery of this small glycopeptide hint at similar molecules involved in the pathology of other diseases by acting as antiproliferative agents? It is tantalizing to say that APF could ignite research in other areas leading to similar discoveries. The difficulties encountered during the isolation of APF (i.e., paucity of material, purification problems, confusion owing to similar activity of different sugar derivatives) may have discouraged similar investigations in other diseases states. Nonetheless, the fascinating basic science behind the APF discovery, along with the possible medical benefits of this novel agent, should encourage others to pursue similar avenues to further this field.

The sequence of Antiproliferative Factor (APF). APF consists of a primarily hydrophobic nonapeptide sequence covalently linked to a trisaccharide. Although a terminal sialic acid moiety is present in the natural product, sialylation is not required for APF’s activity, although the presence of a glycan is critical. The de-sialylated peptide, termed asialo-APF (as-APF), has the same biological activity and nearly the same potency of APF. T, threonine; V, valine; P, proline; A, alanine. Used by permission (9).
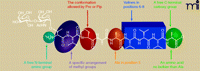
The structural elements important for APF’s activity. Structural features of the peptide portion of as-APF needed for activity as determined by SAR (9). Groups of amino acids are highlighted in colored boxes and a brief statement concerning their importance is annotated. A synopsis of the SAR that led to Figure 2⇓ (determined through the synthesis and biological evaluation of thirty-six analogs) is given in Box 1. Reprinted with permission.
- © American Society for Pharmacology and Experimental Theraputics 2009
References

Piotr Kaczmarek, PhD, earned his doctoral degree in bio-inorganic chemistry at The University of Wroclaw, Poland. He came to the United States to work as a post-doctoral fellow with Christopher Michejda in the Structural Biophysics Laboratory, in the Center for Cancer Research at the National Cancer Institute-Frederick, to work on the chemistry and biochemistry of APF. After Dr. Michejda’s untimely death in 2007, Dr. Kaczmarek has continued his research on APF under the supervision of Joseph J. Barchi, Jr. in the Laboratory of Medicinal Chemistry, CCR, NCI.

Joseph J. Barchi, Jr., PhD, graduated from Rutgers University as a Henry Rutgers scholar, earning an AB in chemistry. He received his doctoral degree in Synthetic Organic and Marine Natural Products Chemistry from the University of Hawaii with Richard E. Moore and held a two-year postdoctoral fellowship at Duke University with Bert Fraser-Reid. In 1988, he joined the National Cancer Institute as a staff fellow in the Laboratory of Medicinal Chemistry (LMC). He currently is a Senior Scientist/Investigator and is the NMR Facility Head at the LMC. His main research interests are in synthetic medicinal chemistry as it relates to carbohydrate-based drug design; the development of novel sugar-conjugated nanoparticles; and the high-resolution structural analysis of sugars, glycopeptides, nucleosides and small molecule drug candidates by NMR spectroscopy. E-mail barchi{at}helix.nih.gov; fax 301-846-6033.

