Parstatin, a Novel Protease-Activated Receptor 1–Derived Inhibitor of Angiogenesis
- 1 Division of Matrix Biology, Department of Medicine, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA 02215
- 2 Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02215
- 3 Harvard-MIT Division of Health Sciences and Technology, Boston, MA 02215
Angiogenesis, the process of forming new blood vessels, is a well-established and clinically relevant feature of a variety of disease states (1). Decades of research demonstrate that the rate of tumor growth and resulting cancer progression is dependent upon the establishment of new blood vessels to supply oxygen and nutrients to proliferating cancer cells (2–4). Additionally, in the wet form of age-related macular degeneration, retinal bleeding that occurs as a direct result of angiogenesis can subsequently lead to scar formation and blindness (5). Whether blood vessels sprout in a given tissue environment depends on the balance between factors that stimulate angiogenesis and those that impede it (6). Potent pro-angiogenic factors such as vascular endothelial growth factor (VEGF) have been identified, validated, and successfully used in the clinic (7). Likewise, anti-angiogenic factors are also emerging as biologically relevant and therapeutically useful entities (8). Known endogenous inhibitors of angiogenesis include a number of growth factors, cytokines, and metabolites, as well as peptide fragments that are derived from tissue remodeling (9). That the mechanisms regulating angiogenesis are complex is suggested by the expanding and eclectic classes of molecules that act as inhibitors, comprising not only direct gene products but, in many cases, derivatives of tissue processing. The class of inhibitors derived from post-translational proteolytic processing includes peptide fragments derived from basement membrane collagens and proteoglycans (10–12), cleaved serum proteins (13, 14), and even a peptide fragment derived from tryptophanyl-tRNA synthetase (15). Given the diversity of molecules that possess anti-angiogenic activity, it perhaps should not be surprising that a cell surface protein gives rise to a new member of the class of anti-angiogenic peptide fragments. In a recent article, Tsopanoglou and colleagues report a novel member of this angiogenesis inhibitor class: parstatin, an N-terminal proteolytic cleavage product from the protease-activated receptor 1 (PAR1) (16).
PAR1 is a G protein–coupled receptor (GPCR) that participates in hemostasis and vascular development (17, 18) and that mediates the angiogenic activity of thrombin (19, 20). PAR1 is expressed on a variety of cell types including vascular smooth muscle cells, endothelial cells, platelets, cardiomyocytes, and a variety of epithelial cancer cell types, such as invasive breast carcinoma and pancreatic adenocarcinoma (21–23). PAR1 is activated through proteolytic cleavage of its first forty-one extracellular residues by a variety of proteases, most notably thrombin (24, 25). The new truncated N terminus serves as a tethered ligand that activates the PAR1 receptor itself. Evidence also exists for intermolecular activation between PAR1 and PAR2, which also requires N-terminal processing (22, 26, 27). However, little effort has focused on the forty-one–residue peptide fragment liberated during PAR1 activation (Figure 1). Tsopanoglou and colleagues have now demonstrated that this peptide has intriguing anti-angiogenic activity, and, in a follow-up study, they demonstrate its potential pharmacological utility using a rat model of ischemic heart disease (16, 28).
A variety of proteases have the ability to release parstatin, including thrombin, activated protein C, and matrix metalloproteinase 1 (MMP1). Full-length parstatin peptide has a prominent hydrophobic domain (residues 1–23) followed by a more hydrophilic domain (residues 24–41). In several in vitro and ex vivo assay systems including endothelial cell tube formation, chick embryo chorioallantoic membrane assay (CAM), and rat aortic ring, parstatin is able to inhibit VEGF- and basic fibroblast growth factor (bFGF)-driven angiogenesis. These assays test for endothelial cell migration/invasion, as well as de novo vessel sprouting, and have therefore become widely utilized to assess the pro- or anti-angiogenic capacity of a molecule. Parstatin also inhibited human umbilical vein endothelial cell (HUVEC) proliferation. It is noteworthy that parstatin was specific in its effects; it inhibited VEGF- and bFGF-driven proliferation [via Erk1/2 (p42/44) activation] but had little effect on epidermal growth factor (EGF)- and heparin-binding EGF (HB-EGF)-stimulated proliferation. Although no other cell types were studied—of particular interest would be non-endothelial cell types that are VEGF/bFGF responsive—the in vitro data suggest some degree of specificity by parstatin on endothelial cell proliferation. Parstatin is also pro-apoptotic and leads to the activation of the caspase-dependent cascade of programmed cell death.
Exogenous parstatin rapidly localizes to the cell surface, penetrates the cell membrane, and accumulates in the intracellular space. These events appear to rely on a hydrophobic sequence at the N terminal, as a truncated version of parstatin lacking this sequence is ineffectual. The mechanism by which parstatin is internalized remains to be established. Studies to determine how parstatin accumulates at the cell surface, including the identification of extracellular receptors, potential associations with the lipid bilayer, and the mechanism of peptide internalization, will facilitate understanding of its mode of action. Also of interest is the mechanism of parstatin’s anti-proliferative activity. Is it acting as a competitive inhibitor of other ligand-receptor interactions? Does parstatin directly inhibit intracellular signaling pathways? Furthermore, is parstatin able to disrupt membrane integrity at the cell surface, nucleus, or other cellular compartment?
Although the biological activity of parstatin appears to be dependent upon the hydrophobic N terminus, conjugation to a more hydrophilic sequence is required to improve the poor aqueous solubility of the N-terminal domain. From a therapeutic perspective, this requirement may be advantageous. The activity of parstatin is not affected by the nature of its hydrophilic C terminus, thus providing opportunities for the design of conjugated peptides with blood vessel targeting or dual action motifs. Such strategies may improve the efficacy and the pharmacodynamic and pharmacokinetic profiles of parstatin.
A follow-up study explored the pharmacological properties of parstatin in a model of cardiac ischemia and reperfusion injury (28). Intravenous injection of parstatin was cardioprotective when administered either prior to or directly following injury. Furthermore, parstatin served as a nitric oxide-dependent vasodilator, acting on myocardial myocytes. These results reveal in vivo biological activity and point to the potential therapeutic value of parstatin for cardiac ischemia.
Although parstatin has attractive pharmacological properties, key questions regarding its physiological role in vivo remain unanswered. Liberation of parstatin is protease-dependent, and several types of proteases are capable of releasing this peptide through activation of the PAR1 receptor (24). Inflammation and induction of the blood coagulation cascade are two physiologic processes associated with high amounts of protease activity that are associated with PAR1 activation (22). These processes are common to a wide variety of disease states including cancer, fibrosis, and arthritis. To what extent is parstatin involved in these processes? Additionally, PAR1 activation has been demonstrated to be pro-angiogenic (20). Does parstatin cleavage (coinciding with PAR1 activation) serve to regulate and counterbalance signaling via its activated parent molecule? The answers to these questions and indeed more will help to elucidate the mechanism of parstatin activity and further establish the role of PAR1, as well as its processing and receptor activity, in numerous physiological and pathophysiological processes.
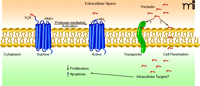
Processing of parstatin from the protease-activated receptor (PAR1). PAR1 is a G protein–coupled receptor expressed on a variety of cell types. Serine proteases activate PAR1 by cleaving a forty-one amino-acid fragment and inducing activation of a tethered ligand. The cleaved peptide, parstatin, localizes at the cell surface and is internalized perhaps by transporter- or lipid-mediated cell penetration. Parstatin subsequently inhibits cell proliferation and induces apoptosis.
Acknowledgments
RK is supported by National Institutes of Health [Grants DK55001, DK62987, AA13913, DK 61688, and CA125550].
- Copyright © 2009
References

Michael Duncan, PhD, is currently a Research Fellow in Raghu Kalluri’s laboratory in the Division of Matrix Biology at Beth Israel Deaconess Medical Center and Harvard Medical School. He received his doctoral degree from the University of North Carolina at Chapel Hill in 2006 and is a 2007 UNCF/Merck Postdoctoral Scholar. E-mail mduncan1{at}bidmc.harvard.edu

Raghu Kalluri, MD, PhD, is a Professor of Medicine at Harvard Medical School and Chief of the Division of Matrix Biology at the Beth Israel Deaconess Medical Center. He received his medical degree from Brown University and his doctoral degree from the University of Kansas. The work in Dr. Kalluri’s laboratory is supported by NIH grants DK55001, DK62987, AA13913, DK 61688, and CA125550, the Champalimaud Foundation and research funds of the Division of Matrix Biology at Beth Israel Deaconess Medical Center E-mail rkalluri{at}bidmc.harvard.edu; fax 617-735-4603.

