Therapeutic Windows
Temozolomide (Temodar®)
Starting from mechanistic studies of alkylnitrosoureas, the serendipitous liaison between university researchers and medicinal chemists culminated in the antitumor drug temozolomide, today an essential element in the arsenal of anti-proliferative, DNA-alkylating agents for the treatment of malignant glioma. The application of temozolomide, alone and in combination, continues to expand.

Discovery (in a University Lab!)
Can university research groups, including their ever-important PhD students, discover new drugs? In a provocative follow-up to his analysis of the reasons for the diminished efficacy of the pharmaceutical industry, Pedro Cuatrecasas firmly proposes so (1). In his appraisal of “Big Pharma,” Cuatrecasas traces problems of diminished creativity and productivity to the obsessive “management” of research that stems from a pervasive fear of unmanageable surprises. He suggests that academic groups, which enjoy a more free-wheeling culture, are more open to the often serendipitous nature of scientific enquiry, and that such groups might indeed do a better job of novel drug discovery. The truth, as usual, lies somewhere in between, as the tale of the discovery of temozolomide illustrates.

Ins and outs
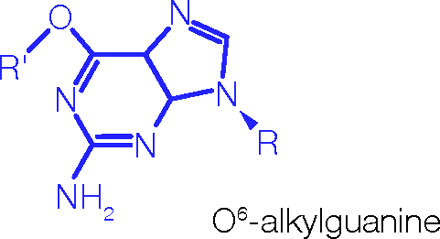
After oral administration, peak plasma concentration is achieved in about 1 hour; food intake interferes with absorption; distribution is on the order of 0.4 L/kg, with weak binding to plasma proteins. Cytochromes P450 play limited role in metabolism; temozolomide is spontaneously hydrolyzed at physiological pH to the active species (see center box). Elimination half-life is approximately 1.8 hours. (See www.spfiles.com/pitemodar.pdf.)
Teaming up for temozolomide
John A. Hickman, Neil Gibson, and Simon Langdon
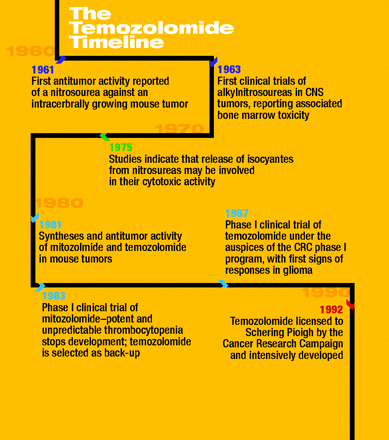
The discovery of temozolomide, at the technical University of Aston, in the industrial city of Birmingham UK, owes a huge debt to the “Cuatrecasian” vision held by the late Professors Tom Connors and Alistair Curry. In the late 1970s, they were able to distribute substantial funds, with minimal peer review, from the Cancer Research Campaign (the UK charity now called CRUK), to kick-start a number of initiatives. They took a risk with a rather young duo of enthusiastic cancer researchers (John Hickman and Andy Gescher) and a medicinal chemist (Malcolm Stevens), with the idea of building an integrated, academic drug discovery group. John Hickman had introduced to Aston a number of mouse tumor models for both in vivo and in vitro use, and dynamic Glaswegian PhD student Neil Gibson, who is currently Chief Scientific Officer of Pfizer Oncology, set about to understand the role that nitrosourea breakdown might play in the activity of this important anticancer drug class. It was a classical study of mechanism of drug action, except that one nitrosourea breakdown product—the isocyanate product—was predicted to modify proteins rather than DNA, indicating a novel mechanism of action and potentially permitting the targeting of tumor biochemistry in an innovative way. Bob Stone, who was supported as a PhD student with Malcolm Stevens by a grant from the UK Science Research Council and the pharmaceutical company May and Baker, was at about the same time pursuing a project based on isocyanate chemistry. He was using isocyanates to synthesize cyclic triazinones, which were of potential interest as anti-allergic agents. It did not take us long to have the idea that these synthetic reactions might be exploited in reverse to generate protein-reactive isocyanates.
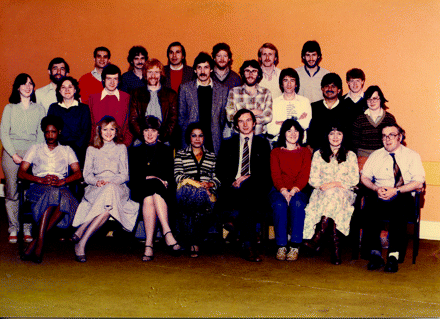
Snapshot (1980s): Some of the Cancer Research Campaign’s Experimental Chemotherapy Group. Center (standing) John Hickman and Andy Gescher (leather jacket); top, far right, Neil Gibson; top, far left, John Slack; middle, third from right, Simon Langdon (white T-shirt); front row, center, Malcolm Stevens (sitting).
We referred to the potential of isocyanate-generating agents, with excitement, in a paper published in 1982 (2). Neil Gibson injected a group of TLX5 lymphoma–bearing mice with a chloroethylisocyanate-based triazinone (CCRG 81010), which in-house was called Azolastone (crossword aficionados may wish to figure that name out). The treatment was completely successful in this model as well as in a range of subsequent in vivo models (3). Mechanism-of-action studies by Len Erickson at the NCI soon showed that the molecule had been effective for unexpected reasons: the isocyanate was not rereleased, as we had predicted, but rather, Bob Stone had synthesized a pro-drug that was converted into a DNA alkylating agent. The extraordinary antitumor activity of this molecule, which alkylates O6 of guanine in DNA, prompted May and Baker, in the UK, and their parent company Rhone-Poulenc, in Paris, to perform extensive structure–activity studies. Working closely with Eddie Lunt and Chris Newton (UK) and François Lavelle and his team (Paris), Simon Langdon, a postdoc at Aston (currently on the faculty at Edinburgh University), performed extensive screening and established optimal scheduling. Azolastone was renamed mitozolomide and entered phase 1 clinical trials. Unfortunately, the drug proved to be potently thrombocytopenic.
Simon Langdon’s screen, however, had discovered that a related compound (CCRG 81045, M&B39831), in which the chloroethyl group of mitozolomide was substituted by a methyl group, had excellent activity (4). This second molecule, called temozolomide (a name invented by Chris Newton), made its way into humans through the particular tenacity of Malcolm Stevens and the team at Aston, with John Slack and Colin Goddard (now at OSI) undertaking the pharmacokinetics. Trials were initiated primarily at Charing Cross Hospital in London, under the calm direction of the late Edward Newlands, and its activity against glioma was readily determined (5). Mechanistic studies showed it to be a very well tolerated pro-drug form of the alkylating agent dacarbazine. A brilliant development program established the drug for treating CNS tumors and more. The molecule passed into the hands of Schering Plough and continues to generate hundreds of millions of dollars in revenues.
So, indeed, blockbusters can be discovered in university laboratories. In the case of temozolomide, development involved both a large multidisciplinary university team (see photograph) and an immediate industrial collaboration for chemistry. Tight collaborations for clinical assessment through the Cancer Research Campaign, initiated by the late Tom Connors with Ken Bagshawe, carefully chaperoned this molecule to success. Temozolomide was discovered through the work of many academic pharmacologists committed to excellence and teamwork, unencumbered by “management.” The success of the drug proves that, in such an environment, PhD students can achieve great things!
Indicated uses
Approved as treatment, in concert with radiotherapy, of newly diagnosed glioblastoma multiforme in adult patients. Also indicated for refractory anaplastic astrocytoma. (See www.cancer.gov/cancertopics/druginfo/fda-temozolomide.)
MI apologizes that space limitations prevent the acknowledgment of many others who have contributed to the drug development story summarized in these pages.
- Copyright © 2009

