|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2012 | Volume
: 18
| Issue : 2 | Page : 238-240 |
| |
A familial deletion 4q syndrome: An outcome of a paracentric inversion
Meena Lall, Ratna Puri, Pushpa Saviour, Ishwar Verma
Center of Medical Genetics, Sir Gangaram Hospital, Rajender Nagar, New Delhi, India
| Date of Web Publication | 8-Sep-2012 |
Correspondence Address:
Meena Lall
Center of Medical Genetics, Sir Gangaram Hospital, Rajender Nagar, New Delhi 110024
India
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.100780

 Abstract Abstract | | |
Chromosome inversions are intra-chromosomal rearrangements formed when the chromosome breaks occur at two places, and in the process of repair the intervening segments are joined in an inverted or opposite manner. Inversions themselves do not appear to cause clinical anomalies, if balanced. Abnormal phenotypes can occur due to gene disruption at the point of breakage and reunion or due to duplication/deficiency recombinants formed during crossover at meiosis. We report a case with familial deletion 4q syndrome in a 1-year-old female child with dysmorphism and congenital abnormalities. The deletion was an outcome of a paracentric inversion 4q31.2q35.2. The deletion was confirmed by fluorescence in situ hybridization using telomeric DNA probes for chromosome No. 4. An attempt was made to correlate the genotype with the phenotype. The father had the same rearrangement with a milder phenotype. The recurrence risk in such cases is high.
Keywords: 4q syndrome, deletion, inversion
How to cite this article:
Lall M, Puri R, Saviour P, Verma I. A familial deletion 4q syndrome: An outcome of a paracentric inversion. Indian J Hum Genet 2012;18:238-40 |
How to cite this URL:
Lall M, Puri R, Saviour P, Verma I. A familial deletion 4q syndrome: An outcome of a paracentric inversion. Indian J Hum Genet [serial online] 2012 [cited 2016 Jun 1];18:238-40. Available from: http://www.ijhg.com/text.asp?2012/18/2/238/100780 |
| Introduction | |  |
Chromosome inversions are intra-chromosomal rearrangements formed when the chromosome breaks at two places, and in the process of repair the intervening segments are joined in an inverted or opposite manner. Inversions themselves do not appear to cause clinical anomalies, if balanced. Abnormal phenotypes can occur due to gene disruption at the point of breakage and reunion or due to duplication/deficiency recombinants formed during crossover at meioses. The incidence of viable recombinants with duplication or deletion was estimated to be 3.8%. [1] Chromosome 4q deletions are very rare. Their incidence was estimated to be 1/100,000. [2] We report a case with familial inversion 4q31.2q35.2 causing deletion 4q syndrome in a 1-year-old female child with dysmorphism and congenital abnormalities.
| Case Report | | |
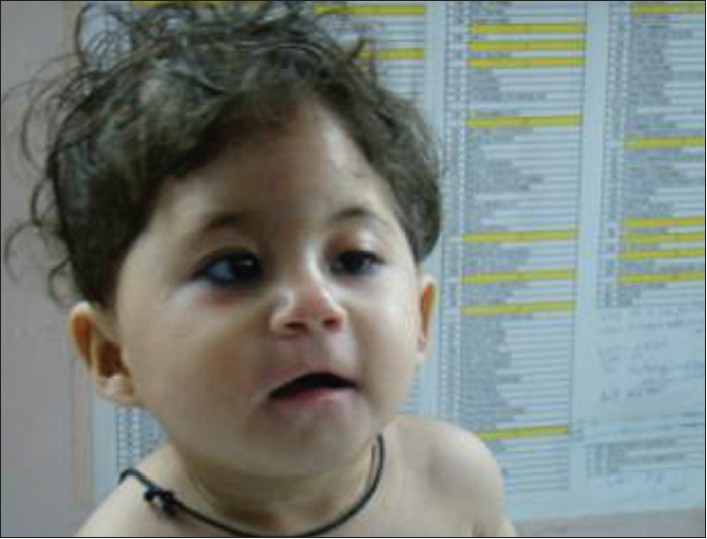
A 1-year-old female child had dysmorphic features [Figure 1] and developmental delay. She was born to a non-consanguineous young couple, with an uneventful pregnancy. The mother was 28 years old and father was 31 years old. The birth weight of the proband was 2.5 kg. She had wide-set eyes, hypertelorism, epicanthic folds, unilateral ptosis, small nose, thin upper lip, low-set ears, and bilateral simian crease, and the thumb was virtually absent, as only a rudimentary spot was present in place of the thumb [Figure 2]. There was no evidence of any gastrointestinal, genitourinary or cardiac defects. Echo of the heart was normal. Analysis of the pedigree of the proposita showed that the father had mild mental retardation and his sister also had a female child with dysmorphism [Figure 3].
Cytogenetic Analysis
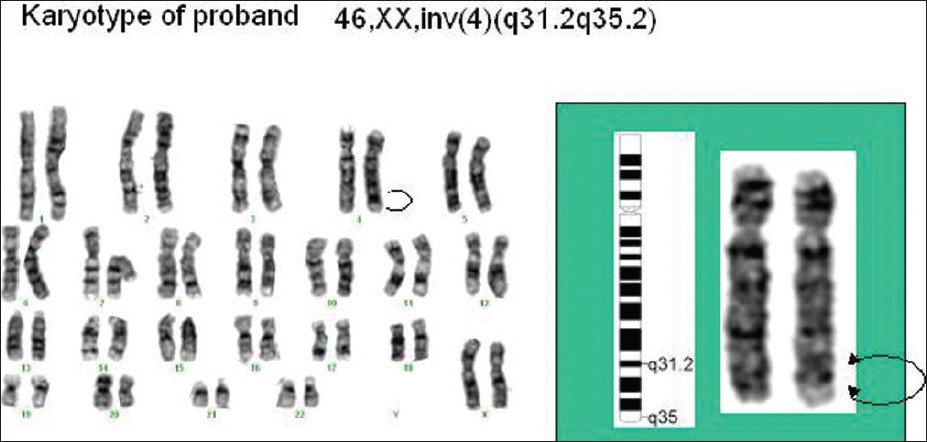
The lymphocytes from peripheral blood were cultured for karyotyping, [3] and GTG banding was done to identify the chromosomes using the nomenclature and chromosome classification of the ISCN, 2009. The karyotype of the proposita was found to be 46, XX, inv (4) (q31.2q35.2) [Figure 4]. To establish the etiology of this aberration, the lymphocytes of both parents were cultured and karyotyped. The karyotype of the mother was 46, XX with no evidence of any chromosomal rearrangement. The father had the same rearrangement as that of the proband and his karyotype was 46, XY, inv (4) (q31.2q35.2).
Molecular-Cytogenetic Analysis
Fluorescence in situ hybridization (FISH) [4] was done on the metaphases from cultured lymphocytes of the proband using subtelomeric DNA probes for chromosome No. 4. The results of FISH [Figure 5] showed that there were two green signals for 4pter and one orange signal for 4qter, denoting that there was deletion of 4qter of one homologue of chromosome No. 4 causing monosomy of 4qter region. The multiple colored DNA probe of chromosome 21 served as control. FISH result from the father's metaphases also showed the presence of deletion at 4qter region.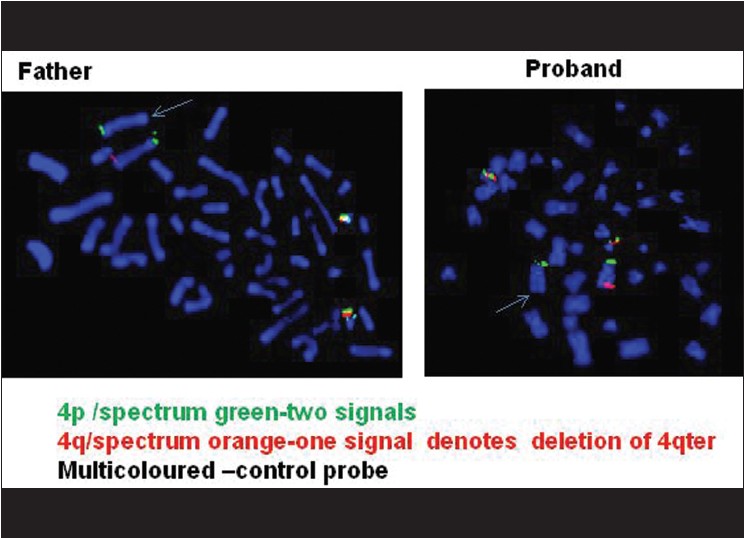 | Figure 5: FISH results with subtelomeric DNA probe of chromosome No. 4 showing deletion of 4qter on one homologue of chromosome No. 4 in the proband and also in the father
Click here to view |
| Discussion | | |
Since the father had the same rearrangement, it was established that the etiology of this aberration was familial. It was not possible to study his sister's child as the family was not willing. The deletion 4q syndrome shows varying phenotype, ranging from mild to severe and complex malformations. Deletion of the terminal region of 4q results in common features including developmental delay, learning difficulties, craniofacial, digital, skeletal, and cardiac anomalies suggestive of a 4q deletion syndrome, as defined by Strehle et al. (2001). [5] The location of the deletion breakpoints vary among patients. The phenotypes of deletions of 4q32 and 4q31 are similar, but more distal deletions at 4q33 and 4q34 are associated with less severe phenotypes. [6] The subtelomeric FISH probe used in the present study showed that the proband and her father had deletion at the subtelomeric distal end at 4q35. There was also no evidence of cardiac anomalies, depicting a milder phenotype compared to other reported cases. [5],[6]
A RefSeq gene in the 4qter region, dHAND, encodes the helix-loop-helix transcription factor that is involved in cardiovascular development and anterior-posterior polarization of the limb bud. Deletion of dHAND has been found in 4q deletion patients with cardiac defects and/or limb abnormalities, but not all patients with dHAND deletions show these phenotypes. [7],[8] Kaalund et al.[6] reported one patient with severe limb abnormalities but no cardiac features and second patient with syndactaly and ventricular septal defect. In our proband, there was presence of limb defect, as the thumb was missing, but there was no evidence of any cardiac anomaly. This reconfirms that haploinsufficiency of dHAND does not necessarily result in cardiac phenotypes but can cause limb defects.
Fulvio et al.[9] reported that with 4q deletion, strabismus, nystagmus, ophthalmoplegia, and optic nerve anomalies have been rarely described in literature previously. In their study, they showed that there is association between optic nerve hypoplasia and progressive external ophthalmoplegia (paralysis of the eye muscle). Unilateral ptosis was observed in the proband of our study.
Facioscapulohumeral Muscular Dystrophy (FSHD) type 1A is associated with the deletion in D4Z4 region of chromosomal tandem repeats. The D4Z4 region is a polymorphic variable number tandem repeat (VNTR) array consisting of 3.3 kb units and is located near the distal end of chromosome 4 at the 4q35 location. Unaffected individuals have on chromosome 4 D4Z4 array that has a span of 11 to 150 contiguous units. In individuals with FSHD, the chromosome 4 D4Z4 repeat array is shortened to a range between 1 and 10 contiguous units. [10] The proband and the father did not complete the criteria of FSHD, so we believe that inversion rearrangement of 4qter in both had not affected the polymorphic D4Z4 region.
To conclude, we found that 4q35 deletion can cause missing digits and paralysis of eye muscles, and is associated with variable severity of the phenotype. Recurrence risk is 50%.
| Acknowledgements | | |
We are thankful to Ms Anju Joshi, Ms Sheweta Pandita, Ms Nitika Sethi for the technical support. The author would like to thank the patient for providing consent to use her photograph in this article.
| References | | |
| 1. | Pettenati MJ, Rao PN, Phelan MC, Grass F, Rao KW, Cosper P, et al. Paracentric inversions in humans: a review of 446 paracentric inversions with presentation of 120new cases. Am J Med Genet 1995;55:171-87. 
[PUBMED] |
| 2. | Quadrelli R, Strehle EM, Vaglio A, Larrandaburu M, Mechoso B, Quadrelli A, et al. A girl with del (4) (q33) and occipital encephalocele: Clinical description and molecular genetic characterization of a rare patient. Genetic Test 2007;11:4-10.
[PUBMED] |
| 3. | Shaffer LG, Slovak ML. An international system for human cytogenetic nomenclature, ISCN. In: Campbell LJ, editors. S Karger, Basel 2009.
|
| 4. | Knight S, Flint J. Mullti-Telomere FISH. In: Fan YS, editor. Molecular cytogenetics: Protocols and applications Tolowa NJ: Humana Press Inc; 2002.
|
| 5. | Strehle EM, Ahmed OA, Hameed M, Russell A. The 4q- syndrome. Genet Couns 2001;12:327-9.
[PUBMED] |
| 6. | Kaalund SS, Moller RS, Tesza A, Miranda M, Kosztolanyi G, Ullmann R, et al. Investigation of 4q deletion in two unrelated patients using array CGH. Am J Med Genet A 2008;146A:2431-4.
|
| 7. | Huang T, Lin AE, Cox GF, Golden WL, Feldman GL, Ute M, et al. Cardiac phenotypes in chromosome 4q -syndrome with and without a deletion of the dHAND gene. Genet Med 2002;4:464-7.
|
| 8. | Vogt J, Ryan E, Tischkowitz MD, Reardon W, Bruetin LA. The tale of a nail sign in chromosome 4q34 deletion syndrome. Clin Dysmorphol 2006;15:127-32.
|
| 9. | Parentin F, Fabretto A, Benussi DG, Petix V, Marchetti F, Dalpra L, et al. Ophthalmic features in a dysmorphic boy with chromosome 4q deletion and duplication. Ophthalmic Genet 2009;30:103-5.
|
| 10. | Mahadevan MS. Exposing a dux tale. Science 2010;24:1607-8.
|
[Figure 1], [Figure 2], [Figure 3], [Figure 4], [Figure 5]
|