|
 
 |
|
REVIEW ARTICLE |
|
|
|
| Year : 2014 | Volume
: 20
| Issue : 2 | Page : 101-119 |
| |
Guidelines for screening, diagnosis and management of hemoglobinopathies
Kanjaksha Ghosh1, Roshan Colah1, Mamta Manglani2, Ved Prakash Choudhry3, Ishwar Verma4, Nishi Madan5, Renu Saxena6, Dipty Jain7, Neelam Marwaha8, Reena Das9, Dipika Mohanty10, Rajendra Choudhary11, Sarita Agarwal12, Malay Ghosh13, Cecil Ross14
1 National Institute of Immunohaematology, Indian Council of Medical Research, K.E.M. Hospital, Parel, Mumbai, India
2 Department of Pediatrics and Chief, Division of Hematology-Oncology, LTMG Hospital, Mumbai, India
3 Department of Hematology, Sun Flag Hospital, New Delhi, India
4 Department of Medical Genetics, Sir Ganga Ram Hospital, New Delhi, India
5 Department of Hematology, Former Head, University College of Medical Sciences, New Delhi, India
6 Department of Hematology, AIIMS, New Delhi, India
7 Department of Pediatrics, Government Medical Hospital, Akola, Maharashtra, India
8 Department of Transfusion Medicine, PGIMER, Chandigarh, India
9 Department of Hematology, PGIMER, Chandigarh, India
10 Department of Hematology, Apollo Hospital, Bhubhaneshwar, India
11 Department of Transfusion Medicine, SGPGI, Lucknow, India
12 Department of Genetics, SGPGI, Lucknow, India
13 Department of Hematology, NRS Medical College, Kolkata, India
14 Department of Hematology, St. John's Medical College, Bangalore, Karnataka, India
| Date of Web Publication | 14-Oct-2014 |
Correspondence Address:
Kanjaksha Ghosh
Director, National Institute of Immunohaematology, 13th Floor, NMS Bldg, KEM Hospital Campus, Parel, Mumbai - 400 012
India
 Source of Support: None, Conflict of Interest: None  | 4 |
DOI: 10.4103/0971-6866.142841

 Abstract Abstract | | |
The β-thalassemias and sickle cell disorders are a major health burden in India. Diagnosis and management of these disorders both in adults and in newborns using appropriate approaches and uniform technology are important in different regions of a vast and diverse country as India. In view of a National Thalassemia Control Program to be launched soon, a need was felt for guidelines on whom to screen, cost-effective technologies that are to be used as well as for establishing prenatal diagnosis programs in regional centers. Newborn screening for sickle cell disorders is in its infancy in India and uniform approaches need to be followed. Also, included are guidelines for monitoring and managing patients who are now growing older and need comprehensive care as well as management of complications of the disease.
Keywords: Diagnosis, guidelines, hemoglobinopathies, management, sickle cell disease, thalassemia
How to cite this article:
Ghosh K, Colah R, Manglani M, Choudhry VP, Verma I, Madan N, Saxena R, Jain D, Marwaha N, Das R, Mohanty D, Choudhary R, Agarwal S, Ghosh M, Ross C. Guidelines for screening, diagnosis and management of hemoglobinopathies. Indian J Hum Genet 2014;20:101-19 |
How to cite this URL:
Ghosh K, Colah R, Manglani M, Choudhry VP, Verma I, Madan N, Saxena R, Jain D, Marwaha N, Das R, Mohanty D, Choudhary R, Agarwal S, Ghosh M, Ross C. Guidelines for screening, diagnosis and management of hemoglobinopathies. Indian J Hum Genet [serial online] 2014 [cited 2016 Aug 23];20:101-19. Available from: http://www.ijhg.com/text.asp?2014/20/2/101/142841 |
| Introduction | |  |
Hemoglobinopathies, which include the thalassemias and structural hemoglobin (Hb) variants, are the most common group of autosomal recessively inherited monogenic disorders of Hb production and pose a significant health burden in India.
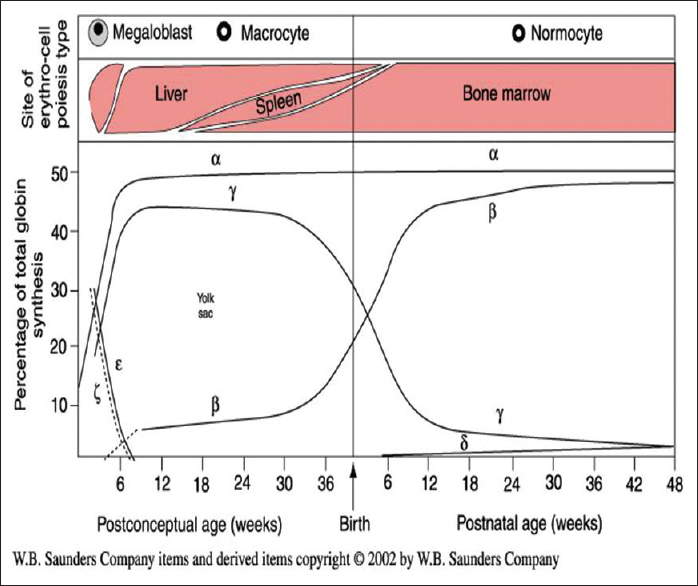
The synthesis of different globin chains of human Hb during development is shown in [Figure 1].
Fetal hemoglobin (Hb F) (α2γ2 ) is the main Hb present during intrauterine life and comprises of 90% of the total Hb up to 34-36 weeks gestation, while Hb A (α2β2 ) represents around 4-13% of total Hb in the fetus. At term, the Hb F levels decrease to 55-90% and the Hb A levels significantly increase to 20-30% of the total Hb. The switch from fetal to adult Hb synthesis continues and by 6 months of age, the amount of Hb F is usually <2-3% and Hb A is the predominant Hb. A small amount of δ globin chains are also synthesized and at birth, the amount of HbA 2 (α2δ2 ) produced is very low but the normal adult levels of HbA 2 (2-3.5%) are usually reached by 6 months of age. [1]
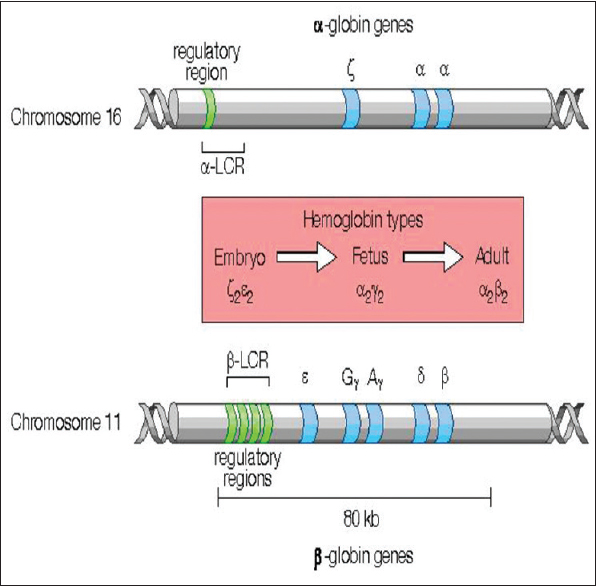
The globin chains are coded by globin genes, which are present in two clusters, the α and α-like globin genes in a cluster on chromosome 16 and the β and β-like globin genes (ε, Gγ, Aγ, δ) on chromosome 11 [Figure 2].
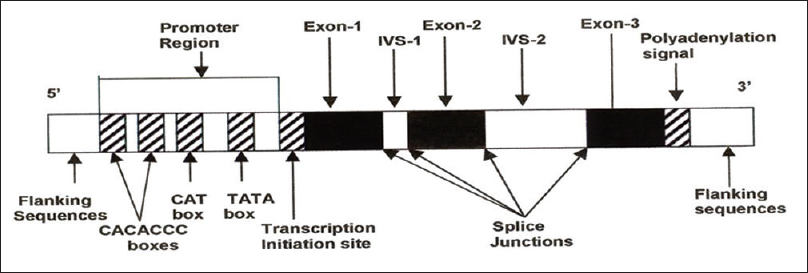
The structure of all the genes is similar and consists of three exons or coding regions, two introns or intervening sequences, a promoter region at the 5′ end before the first exon and a poly A tail at the 3′end [Figure 3]. A locus control region (LCR) located upstream of the cluster and specific promoter sequences regulate the expression of these globin genes.
Mutations in the coding, noncoding, or regulatory sequences in these genes may reduce or abolish the synthesis of α or β globin chains resulting in α or β thalassemia. When mutations in these genes cause structural changes, the structural Hb variants are produced [2] (e.g. Hb S, Hb D, Hb E).
Prevalence of thalassemias and other hemoglobinopathies
The prevalence of β-thalassemia carriers in the Indian population is 3-4%. Some ethnic groups like Sindhis, Kutchis, Lohanas, Punjabis, few Muslim groups as well as few tribal populations have a higher prevalence (5-17%). [3],[4]
The overall prevalence of α thalassemia carriers (single α gene deletion) is around 13% but varies from 3% to 18% in the caste populations, however, it is very high in some tribal groups reaching over 90% in some groups. Hb H disease is uncommon. [5]
δβ thalassemia and hereditary persistence of fetal hemoglobin (HPFH) are less frequent. Among the Hb variants, Hb S carriers are commonly seen in tribal populations as well as in few nontribal groups (5-40%). Hb E carriers are very frequent in the North-East and the prevalence varies between 3% and 64%. Hb D Punjab is mainly seen in the North-Western region with a prevalence of 3-4%. Due to migration and population admixture these Hb variants are now seen all over the country. Hence, the compound heterozygous conditions (Hb Sβ thalassemia, Hb E-β thalassemia, Hb D-β thalassemia) are seen in different regions. [6]
The conditions that need to be identified are summarized in [Table 1]a and b
Individual groups where screening and diagnosis are indicated [7]
- Premarriage screening
- Antenatal screening
- Preconception screening
- Neonatal screening
- Preoperative/preanesthesia screening.
Premarriage screening
This should ideally be done to identify carriers of β thalassemia and the structural hemoglobinopathies like sickle cell trait but is often not acceptable due to social and cultural reasons. Screening can be done in colleges and universities, schools, or community centers. It is particularly relevant in high-risk communities where the prevalence is high.
Antenatal screening
Pregnant women should be screened in all antenatal clinics irrespective of the gestational age, women in the second trimester should also be screened as knowing the carrier status will help in controlling these disorders in subsequent pregnancies if required. Husbands of carrier women with the following Hb abnormalities need to be screened:
- β thalassemia trait
- δβ thalassemia trait
- Hb S trait
- Hb E trait
- Hb D trait
- Hb Lepore trait
- Hb D-βthalassemia
- Hb Q India - β thalassemia.
Prenatal diagnosis should be advised if the fetus is at risk for the following conditions:
- β thalassemia major
- δβ thalassemia major
- β-δβ thalassemia
- Hb S-β thalassemia
- Hb E-β thalassemia
- Sickle cell anemia
- Hb SD disease
- Hb Lepore-β thalassemia
- Hb SE disease.
Screening of antenatal women even later in pregnancy helps to identify couples at risk who could get their babies' blood checked at birth and opt for prenatal diagnosis in subsequent pregnancies.
Preconception screening
Although difficult in the Indian situation, this should be done wherever possible as women most often do not register in antenatal clinics before 12 weeks of gestation. The same approach as for antenatal screening should be followed.
Preconception screening is essential for all couples coming to infertility clinics for assisted conception. If the woman is a carrier of a significant hemoglobinopathy, her partner or sperm donor should be screened and in case of an ovum donor, the donor should also be screened.
Neonatal screening
Newborn screening is mainly recommended for sickle cell disorders among those tribal and nontribal populations where the prevalence of Hb S is high. Ideally, universal screening should be done where all newborn babies in these high risk groups are screened as this would allow identification of other clinically significant disorders such as homozygous β-thalassemia and all cases of Hb S-β thalassemia.
When resources are restricted, a targeted screening approach should be used where the mothers are first screened using inexpensive methods like the solubility test and babies of only those mothers who have a positive solubility test are screened. This approach will miss a few cases of sickle β-thalassemia when the mother is a β-thalassemia carrier and the father is a carrier of Hb S. Other clinically significant hemoglobinopathies will also not be picked up. Babies should be screened either at birth or within the first 4 weeks after birth. All babies with a significant hemoglobinopathy should be re-tested using molecular technology to confirm the diagnosis within 3 months after birth. Babies with sickle cell disease (SCD) or sickle β-thalassemia should be followed-up every 3 months clinically and timely penicillin prophylaxis and pneumococcal vaccination given.
Preoperative/preanesthesia screening
This should be done in patients from ethnic groups where the prevalence of Hb S is high, as the presence of sickle Hb may influence preoperative techniques and clinical management.
Genetic counseling
Genetic counseling should ideally be provided by a medical specialist who has been trained in counseling families with hemoglobinopathies. Counseling can also be given by a trained genetic counselor, a hematologist, or a pediatrician.
Guidelines for laboratory methodology to be followed [7],[8],[9],[10]
Diagnosis of patients with significant hemoglobinopathies
Patient and family history, clinical evaluation, and the origin of the family should be recorded. Full blood count (FBC) on an automated/semi automated hematology analyzer as well as a well stained peripheral blood smear should first be assessed. In cases with microcytosis, iron deficiency anemia and anemia of chronic disease should be ruled out and specific investigations for thalassemia and other hemoglobinopathies considered. The method of choice for Hb analysis is automated cation exchange HPLC. This gives an accurate estimate of HbA 2 , Hb F, and identifies and quantifies the common variant Hbs like Hb E, Hb S, and Hb D Punjab . Hb H is not identified but may give a sharp spike at the start of the chromatogram before 1 min. This instrument is available at many centers in India. Alternative automated methods for Hb analysis are based on isoelectric focusing (IEF) or capillary electrophoresis but these have not been evaluated extensively in India. More recently, mass spectrometry is also being used particularly for identification of variant Hbs.
Screening for identification of carriers of β thalassemia and other hemoglobinopathies
Naked eye single tube red cell osmotic fragility test
This test based on osmotic fragility using 0.36% buffered saline has been used as a preliminary screening test for β-thalassemia carriers extensively in India. In view of the false negative results seen in a small proportion of β-thalassemia carriers, it is not recommended when an automated hematology analyzer is available. If no other facilities are available, Naked Eye Single Tube Red Cell Osmotic Fragility Test (NESTROFT) can be used for preliminary screening for β-thalassemia carriers. Also, since iron deficiency anemia is common in our population, many individuals who are screened would give a false positive result.
Red blood cell indices
Fully automated or semi automated analyzers can be used. Hematology analyzers should be calibrated regularly and controls should be run. Majority of β-thalassemia carriers will have an MCV of <80 fl and MCH of <27 pg with relatively high red blood cell (RBC) counts for the level of Hb. MCH is a more reliable parameter than MCV if the samples are not run within a few hours. Atypical β-thalassemia carriers may have a normal MCV and/or MCH sometimes, and these individuals may be missed while screening. Carriers of variant Hbs like Hb E and Hb S may also have normal indices in 20-30% of cases.
Quantitation of HbA 2 and Hb variants
HPLC gives an accurate estimate of HbA 2 levels and is the method of choice. HbA 2 levels of >4.0% along with reduced RBC indices are indicative of β-thalassemia carriers. HbA 2 levels of >4.0% with normal RBC indices may also be seen in Vitamin B12/folate deficiency, liver disease, and HIV infection. Borderline HbA 2 levels (3.3-3.9%) must be interpreted with caution. Confirmation by DNA methods may be needed for diagnosis in such situations. HbA 2 levels and/or RBC indices may be normal in β-thalassemia carriers with mutations in the promoter region or the poly A tail of the β-globin gene. When the peak in the HbA 2 window is >10%, it is suggestive of another Hb variant. HPLC will also quantitate Hb S, Hb D Punjab and Hb E and some less common variants which may elute in the P 3 window (Hb J variants) or as unknown peaks. Hb Lepore and Hb D Iran elute in the same window as Hb E. In all such cases, a second method should be used like cellulose acetate electrophoresis at alkaline pH, which will help in identification of these variants.
In the presence of Hb S, the HbA 2 levels may be elevated as derivatives of Hb S co-elute with HbA 2 and the measurement of HbA 2 levels will not be accurate.
Capillary electrophoresis is another automated approach for quantitation of HbA 2 . Here HbA 2 and Hb E will elute separately.
For both these techniques, the instruments should be calibrated daily and controls should be run. The HbA 2 peak should elute at a standard time and the HbF window should also be correctly set. The column temperature needs to be adjusted sometimes to ensure this.
IEF has not been extensively validated for quantitation of HbA 2 although it is a good technique for separation of Hb variants.
Cellulose acetate electrophoresis
Electrophoresis of freshly prepared hemolysates on cellulose acetate membranes at alkaline pH followed by elution of the bands for quantitation of HbA 2 is accurate in laboratories that have adequate experience with this technique. However, it is time consuming and cumbersome and is much less used where automated HPLC is available. Densitometric scanning is not recommended for quantitation of HbA 2 .
Cellulose acetate electrophoresis is a cost effective approach for screening for Hb S coupled with the solubility test. Hb H will also be detected as a fast moving band using this technique. Other variant Hbs such as Hb E, Hb Q India , Hb Lepore, Hb D Punjab and HbD Iran will also show altered mobility.
Quantitation of Hb F
Presently HPLC has been the most widely used technique for quantitation of Hb F and it is useful for identification of carriers of δβ thalassemia and HPFH but two minute alkali denaturation can also be used for quantitation of Hb F when Hb F levels are <15%. The latter will underestimate Hb F levels when they are very high. Quantitation of Hb F is useful for identification of carriers of δβ thalassemia and HPFH. The former usually have reduced RBC indices, while the latter have near normal MCV and MCH. However, it is often difficult to differentiate between the two as iron deficiency is also prevalent in India and DNA analysis is then needed. Enumeration of F cells could help to ensure that the peak eluted in the Hb F window is Hb F and not another variant Hb. The Kleihauer test may be used for this and conventionally the pattern of Hb F distribution as pancellular or hetrocellular can be observed. F cell estimation is done more accurately by immunofluorescence or flow cytometry using a monoclonal anti-Hb F antibody.
Screening and identification of α thalassemia
Carriers of α thalassemia can be identified at birth by screening cord bloods for the presence of Hb Bart's, a fast moving Hb on cellulose acetate electrophoresis. HPLC will show a spike at the beginning of the chromatogram but the Hb is not quantitated or identified.
It is difficult to identify α+ and α0 thalassemia carriers (-α/αα) or (--/αα) in adults using hematological investigations. The MCV and MCH levels and RBC counts will be similar to β-thalassemia carriers but the HbA 2 levels will be low or normal. Occasional HbH inclusion bodies may be picked up if the reticulocyte smear is carefully examined. DNA analysis is needed for identification of carriers of α-thalassemia. Hb H disease can be easily identified by the presence of Hb H inclusion bodies and a fast moving band on electrophoresis.
Screening for unstable Hb variants
The heat stability test and isopropanol stability test are used for detection of unstable Hbs. Few variants may show an unknown peak on HPLC or a band with altered mobility on electrophoresis at alkaline pH but many of them show a normal pattern. Since the unstable Hb degrades or denatures readily depending on the location and type of the abnormality, quantitation may not reflect the production of the absolute amount of the abnormal Hb.
Newborn screening
Newborn screening is particularly relevant for sickle cell disorders. Either dried blood spots can be used by taking a heel prick sample on Guthrie cards between 1 and 7 days after birth or cord blood samples can be collected at birth in EDTA. Dried blood spots can be analyzed either by HPLC using the NBS machine from Bio-Rad Laboratories or by IEF. A new system for analysis of dried blood spots by capillary electrophoresis has also been recently launched. Cord blood samples can be analyzed by HPLC using the sickle cell short program specific for newborns or using the β-thalassemia short program used for screening adult samples. The advantage of the latter is that the kit does not need to be changed for adult and newborn samples. All samples where sickle cell anemia, sickle β-thalassemia or Hb SD disease is suspected must be confirmed by molecular analysis before the babies are registered for comprehensive care.
Prenatal diagnosis
Chorionic Villus Sampling (CVS) under ultrasound guidance is done between 10 and 12 weeks of gestation when couples at risk of a severe hemoglobinopathy are identified. The CVS and parental samples are analyzed by one of the mutation detection methods. Usually amplification refractory mutation system (ARMS) and covalent reverse dot blot hybridization (CRDB) are used in India depending on the facilities available at different centers. DNA sequencing is done when the mutations remain unidentified. Variable number of tandem repeat/short tandem repeat (VNTR/STR) analysis should be done to rule out maternal contamination in fetal samples.
For couples who are referred late, as often happensin India, fetal blood sampling (FBS) is done by cordocentesis at 18-20 weeks gestation. The fetal blood can be analyzed directly by HPLC after ensuring that there is no maternal contamination by fetal cell staining using the Kleihauer technique or VNTR/STR analysis. When the percentage of HbA o (adult Hb) in the fetal sample is <1%, mutation analysis must be done to differentiate a heterozygous β-thalassemia fetus from a homozygous β+ or compound heterozygous β+/βo thalassemia fetus. There will be some overlap in HbA 0 levels between heterozygous β-thalassemia and normal fetuses. However, it is recommended that molecular analysis should be done on fetal blood samples as some unusual cases (e.g. homozygosity for the poly A mutation) could give near normal Hb A levels on HPLC.
Guidelines for organization of work at different levels
Primary health centre
Screening for Hb S by solubility test
- This should not be used in children <1 year of age and if a blood transfusion is given in the last 3 months
- In severely anemic patients, the Hb should be adjusted by increasing the amount of blood added
- The reagent should be supplied by the District Hospital
Screening for β-thalassemia should be done at the District Hospital
Primary health centre (PHC) should refer any child with anemia, irritability, poor growth, or organomegaly to the District Hospital
PHCs should have the following medicines: Cap. Amoxicillin, Paracetamol, Folic acid, Cifran, MV/BC, Rantac.
Rural hospital
Facilities for CBC, solubility test and Hb electrophoresis should be available.
Rural hospitals should have the following medicines: Cap. Amoxicillin, Paracetamol, Folic acid, Cifran, MV/BC, Rantac, Inj. Diclo, Inj. Rantac, T. Calcium, T. Roxid, Amoxyclav, T. Penicillin, Inj. 3 rd generation Cefalosporin.
District hospital
Facilities for CBC, solubility test, HPLC analysis, Hb electrophoresis, and serum ferritin estimation, screening for G6PD deficiency, and screening for HIV, HBsAg and HCV should be available. In areas where Hb E is prevalent, facilities for screening for Hb E by the dichloro indophenol DCIP dye test should also be available.
Facilities for ultrasonography, X-ray chest, liver function, and kidney function tests should also be available.
Parental studies should also be done especially if the child is transfused.
If a patient comes in failure and has not been previously transfused, an EDTA and plain blood sample should be collected before transfusion. The sample should be preserved for red cell phenotyping at the tertiary care centre in the blood bank.
The district hospitals should have the following medicines: Cap. Amoxicillin, Paracetamol, Folic Acid, Cifran, MV/BC, Rantac, Inj. Diclo, Inj. Rantac, T. Brufen, T. Penicillin V, Calcium, Roxid, Amoxyclav, Inj. Sodium bicarbonate, Cap. Hydroxyurea.
Facilities for giving blood transfusion, if needed, should be there.
Facilities for vaccination - Pneumococcal, Typhoid, HIB, HepatitisBshould also be available.
Tertiary centre
This will be a medical college where facilities for CBC, HPLC, Hb electrophoresis, and molecular analysis should be available. Prenatal diagnosis should also be done at the tertiary center and hence facilities for CVS, amniocentesis, and cordocentesis need to be established in medical colleges. Facilities for PCR-based diagnosis at least for the common Indian β-thalassemia mutations should be available in medical college.
Referral centre
The referral center would have facilities for DNA sequencing and capillary electrophoresis in addition to all facilities available at the tertiary center. All problematic cases would be referred to this centre for further analysis. Capillary electrophoresis would help to separate some Hb variants, which elute in the same window in HPLC and to quantitate HbA 2 in the presence of HbE. DNA sequencing can also be outsourced to avoid the high cost and maintenance of the DNA sequencer as this facility is now available with many companies at a reasonable cost.
Guidelines for management of patients with b thalassemia [7],[11],[12],[13]
Establish the need for regular blood transfusion
Thalassemia intermedia and Hb E thalassemia patients may not need regular red cell transfusions.
Regular transfusions are also not justified only on the basis of Hb levels. Clinical parameters need to be assessed before putting a thalassemia patient on chronic transfusion therapy because chronic transfusion therapy unless optimized can cause red cell allosensitization, iron overload, viral transmission, etc.
Following parameters suggest that the patient will need chronic red cell transfusions.
- Hb level <7 g/dl on two successive occasions separated by at least 2 weeks (the patient should be on folic acid replacement and there should be no other aggravating cause, i.e. infection, bleeding, etc.)
- Patient's growth, activity, academic performance, zeal, etc., are hampered
- Unnatural bony growth due to marrow expansion
- Development of organ failure such as cardiac failure, edema
- Even if Hb level is >7 g/dl and <10 g/dl, and above clinical features are present, the patient may need chronic transfusion therapy.
Objectives of chronic red cell transfusions
- To ensure adequate Hb level so that O 2 delivery to the tissue is not hampered. This will be indicated by:
- Normal growth spurt
- Increased zeal, energy, enthusiasm, and improved academic purpose
- Improved appetite
- To suppress over active erythropoiesis leading to bone deformities.
Frequency of red cell transfusions
Red cell transfusions should be given at an interval of 2-5 weeks.
This interval is optimized based on:
- The amount of red cells transfused so that pretransfusion Hb remains >9 g/dl but posttransfusion Hb does not go above 12 g/dl
- There is no fluid overload
- Transfusion process is over within a reasonable time (4-6 h)
- Frequency of transfusions is not such that it interferes with patient's normal activities
- Reducing the number of Venepunctures (as lifelong transfusion is needed, peripheral veins need to be preserved well).
Suitable blood products for transfusion
- Red cell concentrates (HCT around 0.65) is suitable
- Leucodepleted (prestorage) blood is desirable
- Thalassemia patients who develop at least two non hemolytic transfusion reactions (NHTRs) and allergic reactions need saline washed red cell concentrates.
How much red cells to be infused?
- Pretransfusion Hb is to be estimated
- Weight of the patient is to be recorded
- If the HCT of the red cell concentrate used is 0.65, then 3-4 ml/kg will raise the Hb by 1 g/dl in the absence of hypersplenism
- Generally in a single transfusion an attempt is made to raise the Hb by 4 g/dl if transfusions are scheduled at 3- to 5 weekly intervals.
[Table 2] summaries the amount of donor RBCs with different hematocrit values, which would need to be transfused to increase the Hb levels in the patient by 1-4 g/dl.
What pretransfusion testing should be done before starting chronic transfusion therapy
- Alloantibbody screening at regular intervals in necessary and ideally it should be done during pretransfusion testing. Once an alloantibody is detected, it should be identified and antibody negative blood should be crossmatched
- Extended phenotyping of patient's red cells (ABO, CcDEe, Fy, Kell) is desirable. It would be desirable to establish an extended phenotyped donor registry
- Confirmation of a diagnosis of thalassemia major or severe thalassemia intermedia
- If the family is interested in stem cell transplantation, then counseling and early referral to such a centre is justified
- Serum ferritin level is not needed if the child is <2 years
- Serum ferritin levels should be recorded at regular intervals (3 months) after first 10 units of red cells have been transfused and iron chelation should be started and the dose should be adjusted so as to maintain serum ferritin between 500 and 1000 ng/ml
- Before starting chronic red cell transfusions, Hepatitis B vaccination should be completed
- Every 3 months, virus serology should be done to detect viral infection at the earliest
- Ideally patient should receive NAT tested blood product
- Detailed record of red cell transfusions should be kept, that is, date, number of units, origin of those units, any complication, management, etc
- All precautions of grouping/crossmatching and transfusion as is applied to any patient on chronic transfusion therapy should be taken
- Close relatives blood should not be transfused.
Chelation therapy
Regular iron chelation should be started once serum ferritin level crosses 500 ng/ml. Both oral and intravenous iron chelators are effective. Two oral iron chelators are available in the market at present.
- Deferiprone (L1)
- Deferasirox (Asunra).
At present, both iron chelators are found to be effective. While Deferiprone increases excretion of chelated iron through urine, Asunra increases excretion through both stool as well as urine.
Deferiprone is given at a dose of 75 mg/kg/day in three divided doses. Asunra is given at 30 mg/kg/day in a single dose after dissolving the tablet in water or orange/apple juice. It has been observed that Indian children require deferiprone in a dose of 100 mg/kg/day while Asunra in a dose closer to 40 mg/kg/day to lower serum ferritin levels.
The action of iron chelators are best on an empty stomach.
The injectable iron chelator is desferrioxamine and it has been used for the longest duration. It is given as a subcutaneous injection over 8-14 h/5 days a week at a dosage of 40 mg/kg/day.
Combination of iron chelation therapy
Desferal and Deferiprone have different sites of action. Both have been used together to improve compliance and efficacy of chelation therapy to reduce the side effects and the cost of therapy depending upon the serum ferritin levels and weight of the child. This combination has been found to be more effective and safe. In several National and International studies, Deferiprone (75 mg/kg/day) is given for 5-7 days in a week and Desferal (30-40/kg/day) is given subcutaneously 2-3 days a week. This combination has been found to be an acceptable regimen. Combination regimen is usually recommended for patients with high serum ferritin levels and patients having cardiac and liver iron overload and in children with endocrine problems. Studies are in progress to evaluate the benefits of combination of Desferal and Deferasirox. The preliminary results have shown that this is more effective and safe. Two oral chelating agents have been used together by few doctors in absence of any studies without any side effects.
Monitoring thalassemia patients
Iron levels of thalassemia patients can be monitored by estimating serum ferritin levels, which is readily available and easy to monitor. When the level is above 1000 ng/ml, chelation therapy should be initiated. Level of serum ferritin above10,000 ng/ml is found to be associated with significant organ dysfunction. The limitation of serum ferritin is that its levels are falsely very high in presence of infection, vitamin C deficiency, hepatic damage, hemolysis, and ineffective erythropoiesis. Till date, serum ferritin still remains the most practical investigation to assess the iron overload. The trends in serum ferritin levels over a period of time (rise or fall) serve as a good indicator of body iron burden and help to adjust the dose of chelation therapy. Other methods to detect iron overload are liver biopsy, magnetic resonance imaging (MRI) and Superconducting Quantum Interference Device (SQUID) and MRI star 2 images of heart and liver. Liver biopsy is the gold standard but it is invasive, expensive, and associated with the risk of internal bleeding. Presently, this is used for research purpose only.
SQUID is an imaging modality and it directly measures the body ferritin and hemosiderin. This facility is not available in India. However, it is not preferred to evaluate the myocardial iron overload.
It is a very good test that indicates iron stored in the heart and liver along with their functions.
MRI provides a noninvasive, quantitative method of measuring tissue iron concentration indirectly. Liver iron levels determined using MRI show excellent correlation with liver iron. MRI has the ability to evaluate the entire organ and offers an accurate method to measure liver iron content. Presently, FerriScan has been developed using MRI, which is a simple and effective method to assess iron overload in different organs and total body iron. Recently T2* MRI is becoming the new standard for measuring cardiac iron levels, it is better than MRI. In general, the lower the T2* value, the higher the risk of cardiac dysfunction, a T2* value below 8 ms is suggestive of severe cardiac iron overload.
Other monitoring
Regular monitoring of thalassemic children is of utmost importance in daycare centers with the following objectives:
- To ensure that pre transfusion Hb is maintained between 9 and 10 g/dl
- To detect development of leucopenia or thrombocytopenia
- To monitor iron status by serum ferritin and MRIT2*
- To detect the side effects of transfusion or complications of chelation therapy early
- Early detection and management of transfusion transmitted infections
- Monitoring of cardiac functions by X-ray chest, Echo, or MRIT2* images if required
- To monitor growth and development and institution of therapy at the earliest for growth failure
- Early detection of endocrine problems and institution of appropriate management.
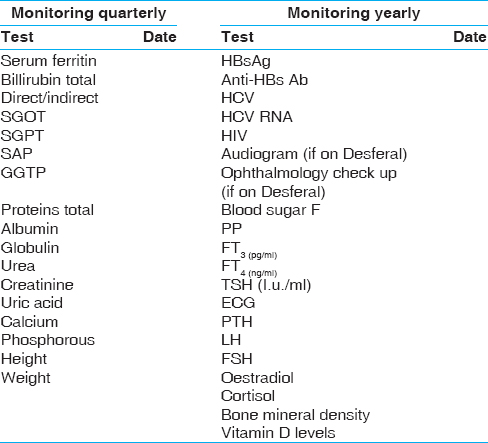
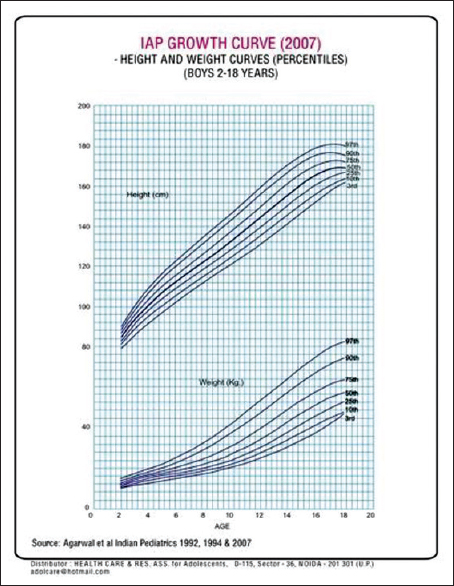
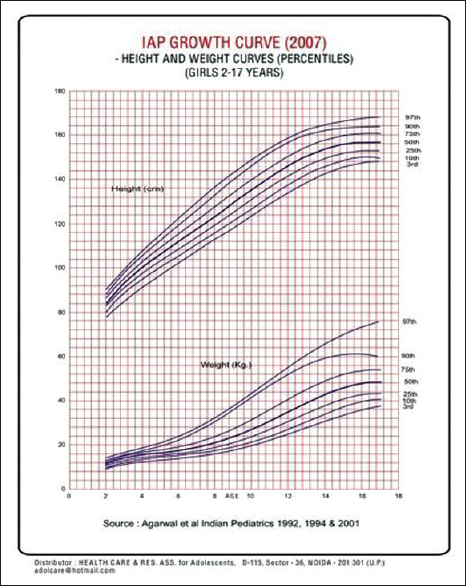
It is desirable that all the clinical and laboratory parameters of the patient should be recorded on a predesigned proforma. Anthropometric measurement should be properly recorded using the growth charts [Figure 4] and [Figure 5]. It will facilitate early detection of growth retardation. Similarly, the serum ferritin levels, calcium and phosphorus levels should be recorded quarterly. Investigations for monitoring of patients at each transfusion are shown in [Table 3] and the tests which need to be done quarterly, annually or after the age of 10 years, are given in [Table 4].
Regular monitoring of thalassemic children is helpful in preventing both complications of the disease as well as of the therapy. It helps in early detection of growth failure, development of endocrinopathies, liver, and cardiac complications. By appropriate and prompt management, most of the complications can be reversed.
Bone marrow transplantation
It offers a permanent cure and better future for children. The credit of the first bone marrow transplantation (BMT) in thalassemia major goes to E Donald Thomas who performed this procedure in an 18-month-old thalassemic child in 1982 using a HLA-matched elder sister as the donor. This child was cured of thalassemia. Since then, many centers in the world and several hospitals in India have initiated BMT facilities.
The principles of BMT include:
- To destroy and prevent regeneration of defective stem cells
- To induce sufficient immune suppression for good engraftment of donor bone marrow cells
- To infuse normal stem cells
- To prevent graft versus host disease (GVHD) with high dose preconditioning, therapy with busulphan, antithymocyte globulin therapy, cyclophosphamide, or total body irradiation. Presently, preconditioning protocols have improved to provide better survival.
The three most important adverse prognostic factors for survival and event-free survival have been observed in large studies, which include:
- Presence of hepatomegaly (hepatomegaly of 2 cm below costal margin)
- Portal fibrosis
- Iron overload (serum ferritin >1000 ng/ml).
Based on these factors, children have been divided into three classes. Class I when all the above three factors are absent. Class II when one or two factors are present and children with presence of all factors are termed as class III. Results of BMT is best in class I children with event-free survival in more than 95% of cases. The cost of BMT in India is around 8-10 lakhs and is regularly being done at Christian Medical College Vellore, Tata Memorial Hospital Mumbai, All India Institute of Medical Science (AIIMS) in New Delhi, Sanjay Gandhi Postgraduate Institute of Medical Science (SGPGI) Lucknow and Post Graduate Institute of Medical Education and Research (PGI) Chandigarh. Several other centers' in corporate hospitals are providing bone marrow transplant facilities.
All thalassemic children should be considered for BMT if a HLA-matched donor is available. Several non government organizations (NGOs) and state governments are providing financial assistance. Cost of BMT is a onetime expense and its results are far better than conventional therapy.
Sickle cell disease
Much progress has been made during the last couple of years in management of SCD. Identification of affected infants provides opportunities for educational and interventions that significantly reduce morbidity and mortality during childhood and adolescence. Comprehensive health maintenance with parental education and appropriate prophylactic measures and monitoring for the development of chronic organ damage are important.
Acute illness in patients with SCD can prove rapidly life threatening; to prevent that, appropriate care must be provided for the management of complications in a setting where knowledge and perspective about SCD is available and where physicians have ready access to baseline information about the patient including results of previous physical examinations, laboratory reports, and radiographs. It is essential that patients have unimpeded access to providers who have the expertise necessary to quickly recognize and treat potentially catastrophic signs and symptoms. Such care not only reduces morbidity and mortality, but it may also reduce medical costs by preventing some manifestations of the disease and by limiting the severity or sequel of others. Many acute complications can be managed safely on an outpatient basis, thus reducing the need for hospitalization.
These guidelines provide information about the diagnosis of SCD, an overview of comprehensive care, and clinical care and protocols for the management of some of the more common acute and chronic complications.
Principles of care for children and adolescents with sickle cell disease
SCD is a complex genetic disorder with multi system manifestations that requires specialized comprehensive care to achieve an optimal outcome. Comprehensive medical care includes ongoing patient and family education, periodic comprehensive evaluations, and other disease-specific health maintenance services, timely and appropriate treatment of acute illness, and genetic counseling. In addition to medical treatment, the management of SCD requires sensitivity to important psychosocial implications of the disease and services to address them. Barriers to appropriate health care include inadequate insurance coverage, transportation, and access to health care provides with expertise in the management of SCD.
Screening of antenatal women even later in pregnancy helps to identify couples at risk who could get their babies blood checked at birth and opt for prenatal diagnosis in subsequent pregnancies.
Comprehensive health care program for sickle cell disease patients [14],[15],[16],[17],[18],[19]
Clinic visit
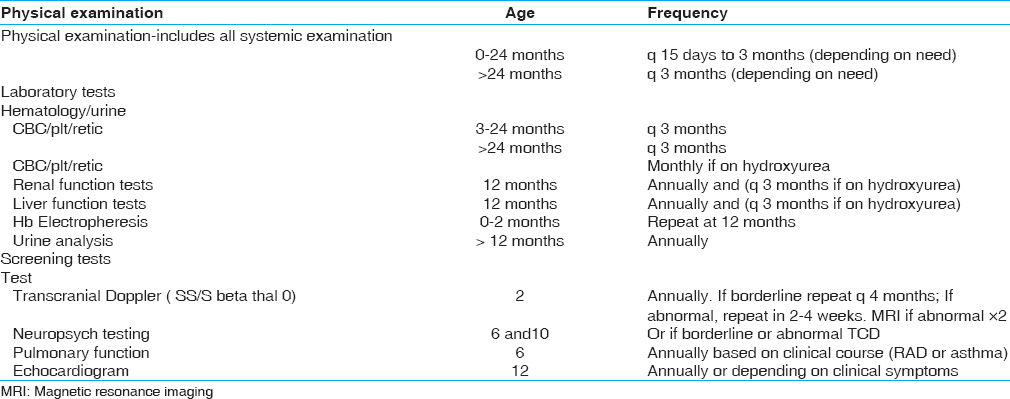
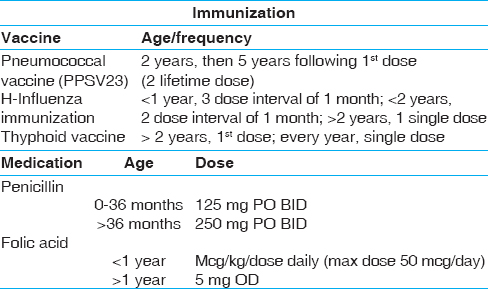
The clinical evaluation and laboratory investigations to be done during the visit of the patient to the clinic are shown in [Table 5]. The immunization and medication schedule is shown in [Table 6].
The age and frequency when referral clinic visits are required are summarized in [Table 7]
Management for sickle cell disease [14],[15],[16],[17],[18],[19]
Acute illness in sickle cell disease
Acute illness requiring immediate medical care, including emergences that need to be defined.
It would include any of the following:
- Temperature >38.5°C
- Pain inadequately relieved by home measures
- Significant respiratory symptoms (e.g. severe cough, shortness of breath, chest pain)
- Abdominal pain, distention, acute enlargement of the spleen
- Any neurologic signs or symptoms even if they are transient
- Significant increase in pallor, fatigue, or lethargy
- Significant vomiting or diarrhea.
Acute illness characterized by any one of the signs or symptoms listed above can prove rapidly life threatening. Thus, it is essential that SCD patients have unimpeded access to the health care providers at the community centers where appropriate measures can be taken. Immediate contact/consultation with a pediatric hematologist or a primary care physician with expertise in SCD is necessary. The patient's baseline data should be available.
Outpatient evaluation and management of febrile illness
One should immediately look for the presentation (e.g. toxic appearance). This should be followed by a physical examination, taking a brief history and systemic examination, laboratory investigations (e.g. CBC, DLC, PLT count and reticulocyte count) should then be done.
Management
- Amoxycillin 250 mg tds should be given and adding Augmentin 40 mg/kg should be strongly considered for severe illness or if CNS infection is suspected. Parenteral antibiotics should be given before other procedures, such as CXR
- Paracetamol 250 mg tds should be given for 3 days
- Summary of patient's last comprehensive evaluation should be available and if not, baseline information should be obtained
- A pediatric hematologist or the patient's primary physician with expertise in SCD must be contacted.
Admission should be strongly considered if one or more of the following criteria are present:
Sepsis, severe anemia, age <1 year in a sickle homozygous or sickle-β thalassemia child, temperature >40°C, signs of toxicity, a patient who has received higher antibiotics, aplastic crisis, severe pain, stroke, acute chest syndrome, splenic sequestration.
Inpatient management of fever
The vital signs should be monitored every 2 h until stable and then 4 hourly. Consider CR monitor and intensive care unit (ICU) for any sign of cardiovascular instability, pulse oximetry for severe illness, or if respiratory signs are present record I+O and weight.
Laboratory investigations
CBC, DLC, PLT count and reticulocyte count should be done initially and daily until there is improvement. This should be compared with the patient's baseline data; blood culture, urine analysis and culture. LFT and KFT must be done and abdominal ultrasound considered for cholelithiasis, cholecystitis or pancreatitis. Blood should be typed and crossmatched if the Hb is 1-2 g/dl or more below the baseline or if there is evidence of acute chest syndrome. An orthopedic consultation may be considered if osteomyelitis or septic arthritis is suspected.
Management
- IV+PO 1-1 ½ × maintenance, increased fluids may be needed if patient is dehydrated or if insensible losses are increased, for example, persistent fever, avoid excessive fluids, which may precipitate or exacerbate acute chest syndrome
- Cefotaxime or cefuroxime 50 mg/kg q 8 h IV should be given and it should be substituted with clindamycin 10 mg/kg IV q 6 h for patients with known suspected cephalosporin allergy. Prophylactic penicillin should be discontinued while the patient is receiving broad spectrum antibiotics. Adding vancomycin 10-15 mg/kg IV q 8 h should be strongly considered for severe febrile illness or for proven or suspected CNS infection or a daily dose of Augmentin 40 mg/kg should be started
- Paracetamol 10-15 mg/kg 6-8 hourly must be given as needed
- Transfusion of RBCs should be considered if the Hb is 1-2 g/dl or more below baseline and the patient shows any signs of cardiovascular compromise
- O2 by facemask, the etiology of a new or increasing supplemental O 2 requirement should be investigated. Unnecessary O 2, which could suppress the reticulocyte count, and exacerbate anemia must be avoided.
For discharge, the patient should be afebrile for >24 h and taking adequate oral fluids, there should be resolution of any pulmonary symptoms and no evidence of anemic crisis. The Hb should be stable and a follow-up possible.
Transfusion therapy for acute complications
RBC transfusions play an important role in treatment of some acute illnesses in patients with SCD. For severe complications, timely transfusions may be life saving. Specific guidelines for the use of transfusions for individual complications are provided throughout these guidelines. In general, appropriate use of red cell transfusions require attention to the following issues:
Indication: Indications for red cell transfusions include acute exacerbation of the patient's baseline anemia that requires increased oxygen carrying capacity, acute life or organ-threatening vaso-occlusive episodes, and preparation for surgical or radiographic procedures.
- Acute exacerbation of baseline anemia:
- Hyperhemolysis
- Hepatic sequestration
- Splenic Sequestration
- Aplastic crisis
- Severe vaso-occlusive events:
- Stroke
- Acute chest syndrome
- Severe infection
- Multiorgan failure
- Preparation for procedures:
- Surgery
- Radiographs with ionic contrast
- General anesthesia.
Selection of transfusion products
Leukocyte-depleted, packed RBCs are recommended and where available minor antigen matched, sickle-negative cells are preferred.
Method
Packed RBC transfusion is appropriate for most situations characterized by acute exacerbation of anemia.
Partial exchange transfusion, generally by erythrocytapheresis, may be needed for severe life-threatening illness or in situations where a relatively high baseline Hb precludes a simple transfusion that would risk hyperviscosity by increasing the Hb level to >10-11g/dl.
Considerations
Simple transfusion with 10 cc/kg of packed RBC typically raises the Hb by about 2 g/dl. Severe anemia that develops over several days may be at risk for volume overload and congestive heart failure from rapid infusion of RBCs. Thus slow correction of the anemia, for example, 4-5 cc/kg packed RBC over 4 h often with furomide or isovolemic partial exchange transfusion may be needed to prevent precipitation of heart failure.
Hyperviscosity
Because sickle red cells are poorly deformable, simple red cell transfusions that increase the Hb levels to >10-11 g/dl may cause hyperviscosity in patients not receiving chronic transfusions and should be avoided.
Management for outpatient painful crisis
The characteristics, location and intensity of pain q 15-30 min should be determined by self-reporting. Pain should also be assessed with a developmentally appropriate pain scale if the patient is familiar with this and understands it. Causes for pain should be assessed and change from baseline in spleen, O 2 saturation and mental status. Previous experience with analgesics must be evaluated if on long acting pain medication at home and PCA started without continuous infusion. Physical examination must include vital signs, pallor, cardiopulmonary status, spleen size, and neurologic symptoms.
Laboratory investigations
CBC, DLC, PLT count, and reticulocyte count must be done initially and daily until there is improvement and compared with patient's baseline data; blood culture is needed if the patient is febrile. Blood type and crossmatch should be done if there is extreme pallor, respiratory or neurologic symptoms or if acute splenic enlargement is present. LFT and KFT, CXR, and pulse oximetry may be needed and abdominal ultrasound may be considered (for cholelithiasis, cholecystitis, pancreatitis).
Management
- Mild-to-moderate pain:
- PCM 10-15 mg/kg daily should be given round the clock for 3 days along with oral fluids
- If there is inadequate relief within 30 min, then codeine I mg/kg po with PCM - 1 tablet tds should be added
- Moderate-to-severe pain
- Diclofinac tab 50 mg - 1 tablet tds and oral fluids must be given
- IV fluids: 10 cc/kg bolus over 1 h should be administered followed by a maintenance rate. Excessive fluids should be avoided unless the patient is judged dehydrated
- Pulse oximetry to be monitored: O 2 to be used by a nasal cannula or facemask.
Management of vaso-occlusive pain (Inpatient)
The vital signs need to be monitored q 4 h and the intake + output recorded, daily weight to be monitored if patient is on parental narcotics and having respiratory symptoms. This is recommended to strongly consider continuous pulse oximetry and consider cardio respiratory monitoring.
Laboratory investigations
CBC, DLC, PLT count and reticulocyte count to be done initially and daily until improving and compared with patient's baseline data blood culture if febrile, type of crossmatch if Hb is 1.5-2 g/dl or more below baseline, respiratory or neurologic symptoms or acute splenic enlargement present, LFT and KFT, CXR if cough, chest pain, hypoxemia, and respiratory symptoms present (patients of VOC are increased risk for acute chest syndrome) and pulse oximetry, consider abdominal ultrasound (for cholelithiasis, cholecystitis, pancreatitis).
Medication
- IV + PO 1 × maintenance. More fluids may be needed if insensible losses are increased (e.g. persistent fever) or to support intravascular volume before transfusion
- Incentive spirometry - 10 breath q 2 h when awake
- Paracetamol 10-15 mg/kg poq 8 h to be given or other antiinflammatory agent if no contraindication is present. If this gives inadequate relief, codeine should be added (PCM + Codeine) or Diclofenac tablets and suppository
- Cefotaxime or cefuroxime 50 mg/kg q 8 h IV to be given. This should be substituted by clindamycin 10 mg/kg IV q 6 h for patients with known suspected cephalosporin allergy. Prophylactic penicillin should be discontinued while patient is receiving broad spectrum antibiotics. Adding vancomycin 10-15 mg/kg IV q 8 h should be strongly considered for severe illness or if large infiltrate with pleural effusion is present
- Pain team consultation should be considered
- O 2 by face mask to be given as needed
- Heating pads should be offered or other comfort measures previously used by the patient and ice and cold packs should be avoided
- Laxatives should be given for constipation
- Analgesics may be weaned as tolerated by decreasing the dose, and not by prolonging the interval between doses. The change of analgesics must be discussed with the patient/family
- Red cell transfusions should be considered:
- Simple transfusion for moderately severe illness, especially if Hb >1 g/dl below the baseline (do not transfuse acutely to Hb >10 g/dl, Hct > 30%)
- Partial exchange transfusion to Hb 10 g/dl and Hb S >30% for severe or rapidly progressive disease (the patient may require transfer to ICU and transfusion medicine consultation for erythrocytapheresis). The femoral or central venous catheters should be removed as soon as possible after the exchange transfusion to reduce risk of thrombosis
- Other clinical care paths to be seen for acute chest syndrome, acute splenic sequestration, aplastic crisis, stroke, priapism if present.
Discharge criteria
The patient should get adequate pain relief on oral analgesics, be afebrile >24 h with negative cultures for >24-48 h. Hb should be stable, the patient should be taking adequate oral fluids and able to take po, follow-up plans coordinated with hematology service. There should be resolution of any pulmonary symptoms with an arranged follow-up.
Acute chest syndrome in a child with sickle cell disease:
Definition
An acute illness associated with lower respiratory symptoms, hypoxemia or new infiltrate on CXR.
History
The following should be noted:
- Hospitalization
- Associated symptoms should be documented
- Intake + output should be recorded and the daily weight taken
- Any oxygen requirement must be reviewed and, baseline oxygen sats determined
- History of asthma, current medication, and drug allergies must be reviewed.
Diagnostics
- CBC, DLC, PLT count and reticulocyte count should be done initially and then daily until there is improvement
- CXR should be done initially and repeated for evaluation of clinical deterioration
- Typing and crossmatching of blood should be considered for severe illness or if the Hb is >1 g/dl below baseline. If available, minor antigen-matched, sickle-negative, and leukocyte depleted RBCs should be kept ready
- A blood culture should be done if the patient is febrile or has history of recent fever
- Blood gas analysis should be considered for severe illness
- Renal and liver function tests are needed for severe illness or if diffuse encephalopathy is present.
- General Care and Treatment: Maintain "euvolemia".IV + p.o. 1--1 ½ - maintenance., more fluid is appropriate only if patient is dehydrated or insensible losses are increased
- Incentive spirometry -10 breath q 2 h when awake
- Encourage ambullation, activity
- Oxygen to pulse oximetry >92% 0or >baseline value if >92%
- Acetaminphen 15 mg/kg po 6 hr or prn T >38 0 C (max dose 60 mg/kg/day)
- Ibuprofen 10 mg/kg po 6--8 hr if no contraindication is present like gastritis, ulcer, coagulopathy, renal impairment). Limit more frequent dosing to 72 hr maximum duration
- Cefotaxime or cefuroxime 50 mg/kg q 8 hr IV.substitute clindamycin 10 mg/kg IV q 6 h for patient with known suspected cephalosporin allergy. Prophylactic penicillin should be discontinued while patient is receiving broad spectrum antibiotics
- Azithromycin 10 mg/kg po first dose then 5 mg/kg qd, erythromycin 10 mg/kg q 6 h po, or other macrolide antibiotic
- Strongly consider adding vancomycin 10--15 mg/kg IV q 8 hr for severe illness or if large infiltrate with pleural effusion present
- Consider one dose of fuosemide 0.5- -1.0 mg/kg IV if sign of fluid overload present
- Consider trial of bronchodilators especially if patient has history of reactive airway disease or wheezing on examination
- Consider positive pressure ventilation
- Consider red cell transfusion:
- Simple transfusion for moderately severe illness, especially if Hb >1 g/dl below baseline (do not transfuse acutely to Hb >10 g/dl, Hct >30%)
- Partial exchange transfusion to Hb 10 g/dl and Hb S >30% for severe or rapidly progressive disease (may require transfer to ICU and transfusion medicine consultation for erythrocytapheresis). Remove femoral or central venous catheters as soon as possible after exchange transfusion to reduce risk of thrombosis
- See other clinical care paths for acute splenic sequestration, aplastic crisis, stroke, priapism if present.
Discharge criteria
Improved pulmonary symptoms, afebrile >24 h and negative cultures for >24-48 h, stable Hb, taking adequate oral fluids and able to take po, medications if applicable, adequate pain relief, follow-up plans coordinated with hematology service, consider follow-up pulmonary function testing and the possibility of chronic transfusions or hydroxyurea.
Acute splenic sequestration
Definition
An acute illness associated with Hb 2 g/dl or more below patient's baseline value with acutely enlarged spleen, mild-to-moderate thrombocytopenia is often present. Reticulocytosis equal to or greater than baseline is usually present. If reticulocyte count is decreased, coexistent aplastic crisis should be considered.
Monitoring
Hospitalize, consider ICU admission, vital signs q 2 h until stable, then q 4 h, CR monitor, continuous pulse oximetry, record I+O, daily weight, serial exams to reassess cardiovascular status and spleen size.
Diagnostics
CBC, Blood culture, USG, CXR, type and crossmatch RBCs stat.
Medication
- IV + PO 1 × maintenance, more fluids may be needed if insensible losses are increased (e.g. persistent fever) or to support intravascular volume before transfusion
- Incentive spirometry 10 breath q 2 h when awake if on parenteral narcotics
- RBC transfusion 10 cc/kg for Hb <4-5 g/dl and signs of cardio vascular compromise. Transfusion may be needed for Hb <7-8 g/dl for patients with relatively high baseline Hb levels (e.g. Hb SC disease). In severe cases, urgent initiation of transfusion prior to inpatient admission may be life-saving. A posttransfusion Hb level <8-9 g/dl is generally recommended to avoid the risk of hyperviscosity that may occur several days later when RBCs sequestered in the spleen may return to the circulation and increase the Hb 1-2 g/dl above posttransfusion levels
- Cefotaxime or cefuroxime 50 mg/kg q 8 h IV if febrile, substitute clindamycin 10 mg/kg IV q 6 h for patient with known suspected cephalosporin allergy. Strongly consider adding vancomycin 10-15 mg/kg IV q 8 h for severe febrile illness or for proven or suspected CNS infection
- If applicable, continue Prophylactic penicillin. Prophylactic penicillin should be discontinued while patient is receiving broad spectrum antibiotics
- O 2 by nasal cannula or face mask if needed to keep pulse oximetry >92% or patient's baseline value, if >92%. The etiology of a new or increasing supplemental O 2 requirement should be investigated
- Acetamonophen 15 mg/kg po q 6 h (max daily dose 60 mg/kg) and ibuprofen 10 mg/kg po q 8 h for any fever and mild pain (Hyperthermia may exacerbate cardiovascular compromise with severe anemia)
- See other clinical care paths for vaso-occlusive pain, acute chest syndrome, aplastic crisis, stroke, priapism if present
- If more than two sequestration crisis, child >5 years, Pnemococcal prophylaxis followed by splenectomy.
Discharge criteria
Stable Hb, adequate pain relief on oral analgesic, afebrile >24 h and negative cultures for >24-48 h, taking adequate oral fluids and able to take po, follow-up plans coordinated with hematology service, resolution of any pulmonary symptoms with arranged follow-up.
Acute stroke or neurologic event
Stroke is defined as an acute clinically apparent neurological event and occurs in 8-11% of children with Hb SS. The common presenting symptoms and signs include hemiparesis, monoparesis, aphasia or dysphasia, seizures, and cranial nerve palsy. Acute neurologic symptoms require urgent evaluation and treatment.
Hospitalize, consider ICU, CR monitoring, vital signs, neurological checks q 2 h.
Diagnostics
Document duration of acute symptoms, any prior neurologic symptoms or trauma, type of crossmatch for transfusion, CBC, DLC, PLT count and reticulocyte count initially and as clinically indicated, coagulation profile, blood and urine culture, MRI and MRA (if MRI/MRA not available, CT without contrast to exclude intracranial hemorrhage, initiation of transition therapy should not be delayed by arrangements for imaging studies), consider CSF culture if febrile and no contraindication present.
Management
- IV + PO at 1 × maintenance
- Partial exchange transfusion or erythrocytapheresis to Hb 10 g/dl and Hb S (patient's RBC) <30%. Remove femoral or central venous catheter as soon as possible after exchange transfusion to reduce risk of thrombosis
- Simple transfusion with RBCs to Hb approximately 10 g/dl may be considered as an alternative to partial exchange transfusion for stable patients with Hb 6-7 g/dl (do not transfuse acutely to Hb >10 g/dl, Hct >30%)
- Rx seizures if present
- Treatment increased intracranial pressure if present
- O 2 by nasal cannula or face mask if needed to keep pulse oximetry >92% or patient's baseline value, if >92%. The etiology of a new or increasing supplemental O 2 requirement should be investigated
- Cefotaxime or cefuroxime 50 mg/kg q 8 h IV if febrile, substitute clindamycin 10 mg/kg IV q 6 h for patient with known suspected cephalosporin allergy. Strongly consider adding vancomycin 10-15 mg/kg IV q 8 h for severe febrile illness or for proven or suspected CNS infection
- If applicable, continue Prophylactic penicillin. Prophylactic penicillin prophylaxis should be discontinued while patient is receiving broad spectrum antibiotics
- See other clinical care paths for pain, acute chest syndrome, aplastic crisis, stroke, priaprism if present
For discharge clinically and neurologically stable >24 h after transfusions, afebrile >24-48 h with negative culture, taking adequate oral fluids and able to take po medications if applicable, follow-up plans coordinated with hematology, rehabilitation, physical therapy, and for Hydroxyurea.
Hydroxyurea protocol
Higher levels of Hb F and lower leukocyte counts are thought to be beneficial in patients with SCD and can be achieved with daily oral administration of hydroxyurea.
The clinical course of each patient with SCD should be regularly reviewed by a pediatric hematologist/sickle cell program and the possibility of hydroxyurea treatment considered.
Indication for hydroxyurea therapy
- Admission for three or more severe vaso-occlusive crisis pain event/year
- Admission for three or more blood transfusions/year
- CNS manifestations
- Acute chest syndrome
- Avascular necrosis of bone.
Exclusion criteria
- Pregnancy
- Inability to comply with daily dosing and frequent lab monitoring.
Dose
10 mg/kg fixed dose.
Monitoring should be done as follows:
- CBC every month with a watch on the plt count
- Reticulocyte count
- LFT, KFT every 3 months
- HPLC for HbF levels.
| Conclusions and Recommendations | | |
- Screening for identification of carriers of hemoglobinopathies should be done before marriage or after marriage before conception or during the early antenatal period
- Neonatal screening is important for sickle cell disorders and should be done among the tribal and nontribal population where the prevalence of the sickle gene is high
- Preoperative screening should also be done in communities where the sickle gene is prevalent
- Measurement of red cell indices and quantitation of HbA 2, Hb F and other variant Hbs are recommended for diagnosis of β-thalassemia carriers and heterozygotes of other Hb variants using automated HPLC. Capillary electrophoresis is also emerging as a suitable technique for screening and can be used
- Facilities as defined should be available at different levels staring with the Primary Health Centre, Rural Hospital, District Hospital, Tertiary Centre and finally the Reference Centre
- Centers managing β-thalassemia patients must first establish the need for transfusion, determine the amount of the appropriate blood product required and the frequency of red cell transfusions
- Pretransfusion testing, which includes extended phenotyping of the patients red cells, testing for viral infections, and estimation of serum ferritin as indicated should be done once chronic transfusion therapy has to be started. Hepatitis B vaccination should also be given and detailed records maintained
- Iron-chelation therapy should be started after the serum ferritin levels cross 500 ng/ml and patients should be monitored regularly
- A comprehensive health care program for children and adolescents with SCD should include all systemic examination, Hb analysis and biochemical investigations as indicated and immunization as per the schedule mentioned
- Both outpatient and inpatient management should be available for management of acute illness and other complications as mentioned in these guidelines.
| References | | |
| 1. | Verma IC, Saxena R, Kohli S. Past, present and future scenario of thalassemic care and control in India. Indian J Med Res 2011;134:507-21.  [ PUBMED]  |
| 2. | Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: An increasing global health problem. Bull World Health Organ 2001;79:704-12. |
| 3. | Madan N, Sharma S, Sood SK, Colah R, Bhatia HM. Frequency of β-thalassemia trait and other hemoglobinopathies in northern and western India. Indian J Hum Genet 2010;16:16-25. [ PUBMED] |
| 4. | Mohanty D, Colah RB, Gorakshakar AC, Patel RZ, Master DC, Mahanta J, et al. Prevalence of β-thalassemia and other haemoglobinopathies in six cities in India: A multicentre study. J Community Genet 2013;4:33-42. |
| 5. | Nadkarni A, Phanasgaonkar S, Colah R, Mohanty D, Ghosh K. Prevalence and molecular characterization of alpha-thalassemia syndromes among Indians. Genet Test 2008;12:177-80 |
| 6. | Colah RB, Gorakshakar AC. Structural hemoglobinopathies. In: Lokeshwar MR, Shah NK, Agarwal B, Suchdeva A, editors. IAP speciality series on Pediatric Hematolgy and Oncolgy of Indian Academy of Pediatrics; 2006. p. 151-61. |
| 7. | Ryan K, Bain BJ, Worthington D, James J, Plews D, Mason A, et al. Significant haemoglobinopathies: Guidelines for screening and diagnosis. Br J Haematol 2010;149:35-49. |
| 8. | Bain BJ. Hemoglobinopathy diagnosis. 2 nd ed. Oxford: Wiley Blackwell; 2006. ISBN: 979 - 1 - 4051-3516-0. |
| 9. | Lewis S, Bain B, Bates I. Dacie and Lewis practical hematology. 10 th ed. UK: Churchill Livingstone; 2006. |
| 10. | Mohanty D, Colah R, editors. Laboratory Manual for screening diagnosis and molecular analysis of hemoglobinopathies and red cell enzymopathies. 1 st ed. Mumbai: Bhalani Publishing House; 2008. |
| 11. | Guidelines for the Clinical Management of Thalassemia. Thalassemia International Federation, April 2000. |
| 12. | Management of Thalassemia. Indian Association of Pediatrics Guidelines; 2006 p. 169. |
| 13. | Vichinsky E, Levine L, Bhatia S. Standards of Care Guidelines for Thalassemias-2008, Children's Hospital and Research Centre, Oakland, CA, USA. |
| 14. | National Institutes of Health, National Heart, Lung and Blood Institute, The Management of Sickle Cell Disease. Chap. 5, 4 th ed. In Child Health Care Maintenance. NIH Publication No. 02-2117; Rev 2002. |
| 15. | Driscoll CM. Sickle Cell Disease. Pediatr Rev 2007;28:259-68. |
| 16. | Recommended Immunization Schedules for Children and Adolescents - United States. Am Acad Pediatr Committee on Infectious Diseases-2008 Pediatrics 2008;121:219-220. |
| 17. | Ambrusko SJ, Ginawardena S, Sakara A, Windsor B, Lanford L, Michelson P, et al. Elevation of tricuspid requrgitant jet velocity, a marker for pulmonary hypertension in children with sickle cell disease. Pediatr Blood Cancer 2006;47:907-13. |
| 18. | Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004;350:886-95. |
| 19. | Sickle Cell disease in childhood: Standards and Guidelines for clinical care. 2 nd ed. 2010 NHS. Sickle Cell and Thalassemia Screening Programme and Sickle Cell Society, UK. |
[Figure 1], [Figure 2], [Figure 3], [Figure 4], [Figure 5]
[Table 1], [Table 2], [Table 3], [Table 4], [Table 5], [Table 6], [Table 7]
|