|
 
 |
| CASE REPORT |
|
| Year : 2014 | Volume
: 9
| Issue : 1 | Page : 49-51 |
|
Charcot-Marie-Tooth disease type 5: A clinical and electrophysiological study
Moawia Elbalal Mohammed1, Salih Mohamed Alawi Albasseri2, Izzadin Elawad3
1 Department of Internal Medicine, Faculty of Medicine, University of Gezira, Wad Madani, Sudan
2 Department of Medcine, Sudan Medical Specialization Board, Khartoum, Sudan
3 Department of Internal Medicine, Faculty of Medicine, University of Sinnar, Sennar, Sudan
| Date of Web Publication | 13-Nov-2014 |
Correspondence Address:
Moawia Elbalal Mohammed
Department of Medicine, Faculty of Medicine, University of Gezira, Wad Medani
Sudan
  | Check |
DOI: 10.4103/1858-5000.144667 
A young male presented to our hospital with a long standing history of spastic paraparesis with no cerebellar or sensory ataxia. He had no sensory level or sphencteric disturbances. There is no similar family history. He had normal magnetic resonance imaging of the cervical and dorsal regions. Electrophysiologic studies including electromyography and nerve conduction were consistent with Charcot-Marie-Tooth disease type 5. This case is being reported for its rarity.
Keywords: Charcot-Marie-Tooth type 5, electromyography, nerve conduction studies
How to cite this article:
Mohammed ME, Alawi Albasseri SM, Elawad I. Charcot-Marie-Tooth disease type 5: A clinical and electrophysiological study. Sudan Med Monit 2014;9:49-51 |
| Introduction | |  |
Charcot-Marie-Tooth (CMT) disease, also called hereditary motor and sensory neuropathy, is a common genetic disorder of peripheral neuropathy with an incidence of about 1 in 2500 persons. [1] The clinical phenotypes of all forms of CMT are generally similar and manifest as distal muscle weakness with a revered champagne bottle, pes cavus, peroneal muscle wasting, claw hands with intrinsic hand muscle wasting, a decrease or absence of tendon reflexes, and sensory impairments. [2]
The disease is really a spectrum of disorders caused by a specific mutation in one of the several myelin genes that result in defects in myelin structure, maintenance, and formation. Each gene locus serves a specific function in maintaining myelin integrity. In addition to gene mutation, the clinical expression of these conditions is affected by "gene dosage imbalance" caused by DNA duplications or deletions. [3],[4] Interestingly, different mutations in the same gene may present different neuropathies, whereas the same phenotype may be associated with different gene mutations. [5] Therefore, molecular genetic studies, as well as clinical features and electrophysiological and pathological studies are very important. Recently, we encountered a patient with a peripheral neuropathy, which was consistent with CMT type 5. In this report, clinical manifestations and electrophysiological analyses were studied.
| Case report | | |
A 25-year-old man reported to our hospital with a 5-year history of an intermittent neck pain, radiating to the tip of his both shoulders. The pain is not restricting his movements. Then, after a while, he noticed heaviness and weakness of his both lower limbs, to the extent that they got stuck-up over each other during walking. He had no headache, convulsions or symptoms suggesting cranial nerves involvement. He had no sensory impairment, and his sphincters were intact. The whole scenario took place while he was 21 years of age, and none of his family members had a similar condition.
Clinical examination revealed a young tall man, who is not pale, jaundiced or cyanosed. His blood pressure was 110/70 mmHg, and his pulse was 75/min. His cardiovascular system, chest and abdomen were normal. During the examination of his central nervous system, he was found to be fully conscious, oriented for time, place, and person. He had a normal memory and average intelligence. His speech and cranial nerves were normal. Fundal examination showed no abnormalities. Examination of his upper limbs showed wasting of first interosseous muscles of his both hands; he had no deformity or fasciculations. The power, tone, reflexes, and sensation were all normal.
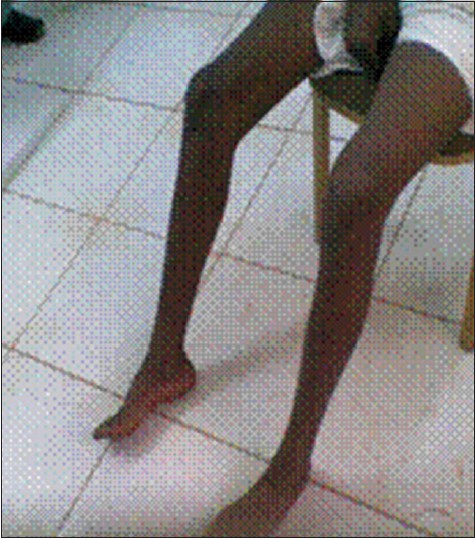
Both of his lower limbs were laterally rotated with wasting of muscles of his both thighs, calves, and feet, giving rise to the typical inverted champagne appearance [Figure 1]. There is pes cavus. Lower limbs were spastic, with brisk reflexes, ankle clonus, and upgoing planters. His sensations were so far intact, and his gait was spastic.
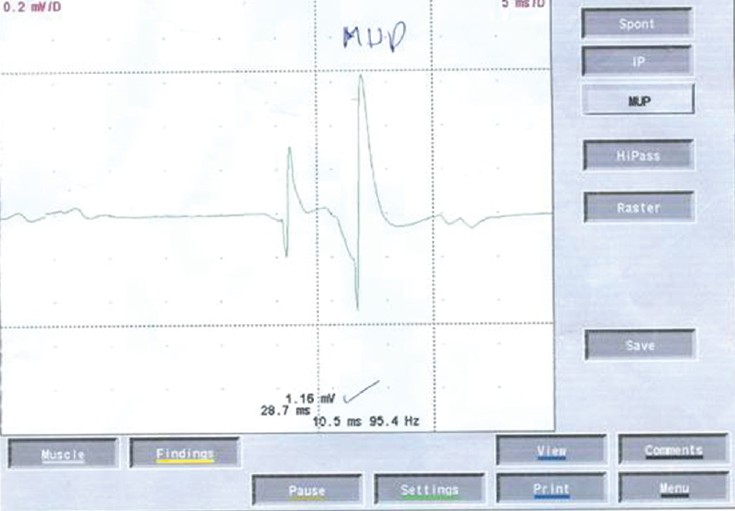
Laboratory studies revealed a normal hematocrit, hemoglobin, white blood cells count, differential, platelet, and red blood cell morphology. Erythrocyte sedimentation rate was 20 mm/h, liver function tests, renal function tests, and lipid profile were all within normal limits. Stool and urine examination were normal. His chest X-ray was normal. Examination of his cerebrospinal fluid showed: A protein of 29 mg/dl, sugar of 49 mg/dl, a negative ZN stain, and a negative Gram-stain for other bacteria. Magnetic resonance imaging of his both cervical and dorsal spine were all normal. Nerve conduction study showed that he has very prolonged distal motor latencies and to a lesser extent proximal latencies, but he kept nearly normal velocities. He has very low amplitude of compound muscle action potentials with waves. The sensory velocity of the peroneal nerve was decreased to 34 m/s [Figure 2]. Electromyography sampling revealed mild chronic denervation features. These results are consistent with a diagnosis of hereditary motor and sensory neuropathy type 5, which is, usually, associated with spastic paraparesis [Figure 3]. Since then he could only be helped by multivitamins and physiotherapy. The patient was carefully followed-up for 1 year, and his clinical course slowly progressed to become bed ridden.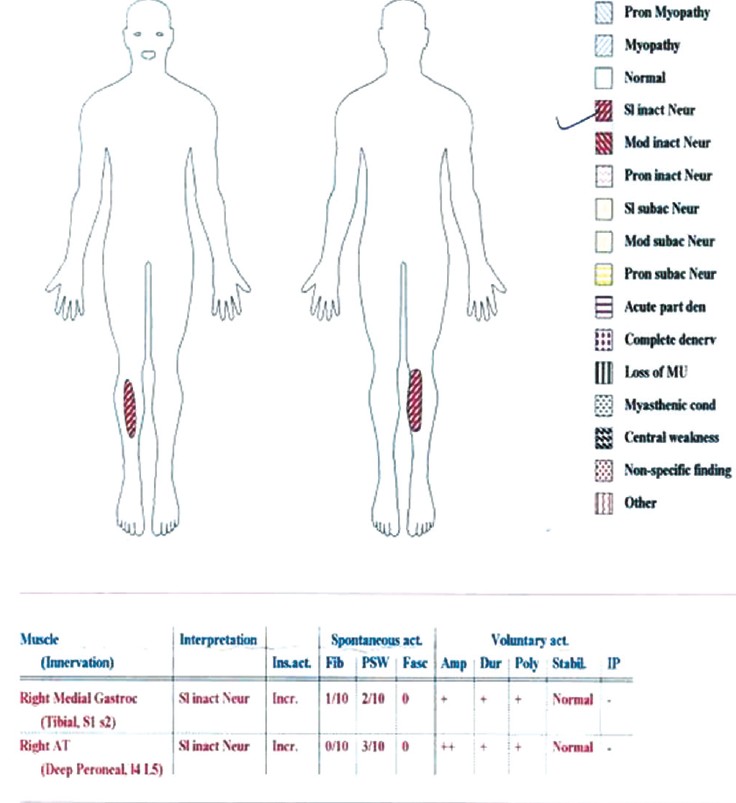 | Figure 2: Nerve conduction study showing a decreased peroneal nerve velocity
Click here to view |
 | Figure 3: Electromyography showing chronic inactive denervation features
Click here to view |
| Discussion | | |
The present study shows the clinical features with a typical demyelinating nature in electrophysiology in a young man with CMT-5.
A diagnosis of CMT-5 depends on autosomal dominant inheritance, clinical manifestations, nerve conduction velocity (NCV) studies, nerve pathology, and even genetic studies. The clinical manifestations include a spastic paraparesis, high arches, and hammertoes with a predominantly axonal motor sensory neuropathy. The disease has a fairly good prognosis, but the prominent weakness and spasticity may cause disability later in life. [6]
In CMT-5 patients, NCV studies, usually, show a homogeneous reduction in NCV in the common peroneal nerve. [7],[8] Our patient has prolonged distal latencies, and reduced conduction velocity down to 34 m/s which are compatible with the typical presentation. The disease may be caused by mutation in MFN2 and BSCL2 (associated with several phenotypes: CMT-2, Dhmn, Silver Syndrome [spastic-paraplegia with hand amyotrophy] and pure spastic-paraplegia). [6],[9]
Because we do not have means of doing histological (nerve biopsy) and a molecular genetic study in our hospital, the diagnosis was clinched by electrophysiological studies. Although molecular genetic studies are very efficient, rapid, and accurate in making a diagnosis, some CMT patients do not have specific responsible genes. Therefore, we conclude that clinical manifestation, electrophysiological findings, and neuropathological studies, as well as molecular genetic studies, remain crucial for a clinical diagnosis.
| References | | |
| 1. | Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet 1974;6:98-118.  [ PUBMED] |
| 2. | Garcia CA. A clinical review of Charcot-Marie-Tooth. Ann N Y Acad Sci 1999;883:69-76. [ PUBMED] |
| 3. | Lupski JR. Charcot-Marie-Tooth polyneuropathy: Duplication, gene dosage, and genetic heterogeneity. Pediatr Res 1999;45:159-65. [ PUBMED] |
| 4. | Kamholz J, Menichella D, Jani A, Garbern J, Lewis RA, Krajewski KM, et al. Charcot-Marie-Tooth disease type 1: Molecular pathogenesis to gene therapy. Brain 2000;123:222-33. |
| 5. | Mersiyanova IV, Ismailov SM, Polyakov AV, Dadali EL, Fedotov VP, Nelis E, et al. Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients. Hum Mutat 2000;15:340-7. |
| 6. | Bertorini T, Narayanaswami P, Rashed H. Charcot-Marie-Tooth disease (hereditary motor sensory neuropathies) and hereditary sensory and autonomic neuropathies. Neurologist 2004;10:327-37. |
| 7. | Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol 1968;18:603-18. |
| 8. | Gilliatt RW, Thomas PK. Extreme slowing of nerve conduction in peroneal muscular atrophy. Ann Phys Med 1957;4:104-6. [ PUBMED] |
| 9. | Espay A. Neuromuscular disorders. In: Espay A, Biller J, editors. Concise of Neurology. Philadelphia: Lippincott Williams and Wilkins, A Wolters Kluwer Business; 2011. p. 283. |
[Figure 1], [Figure 2], [Figure 3]
|








 Search Pubmed for
Search Pubmed for