

Pharmacogenomics of Mood Stabilizers in the Treatment of Bipolar Disorder
- Alessio Squassina squassina{at}unica.it1
- Mirko Manchia mirkomanchia{at}unica.it1,2
- Maria Del Zompo delzompo{at}unica.it1,3
- 1Laboratory of Molecular Genetics, Unit of Clinical Pharmacology, Department of Neuroscience “B.B. Brodie”, University of Cagliari, sp8 Sestu-Monserrato, km. 0,700, Monserrato 09042, Cagliari, Italy
- 2Department of Psychiatry, Dalhousie University, 5909 Veterans Memorial Lane, Halifax, Nova Scotia, Canada, B3H 2E2
- 3Unit of Clinical Pharmacology, University Hospital of Cagliari, Via Ospedale 46, 09124 Cagliari, Italy
Abstract
Bipolar disorder (BD) is a chronic and often severe psychiatric illness characterized by manic and depressive episodes. Among the most effective treatments, mood stabilizers represent the keystone in acute mania, depression, and maintenance treatment of BD. However, treatment response is a highly heterogeneous trait, thus emphasizing the need for a structured informational framework of phenotypic and genetic predictors. In this paper, we present the current state of pharmacogenomic research on long-term treatment in BD, specifically focusing on mood stabilizers. While the results provided so far support the key role of genetic factors in modulating the response phenotype, strong evidence for genetic predictors is still lacking. In order to facilitate implementation of pharmacogenomics into clinical settings (i.e., the creation of personalized therapy), further research efforts are needed.
1. Introduction
Bipolar disorder (BD) is a clinically severe psychiatric disorder, characterized by recurrent alternating episodes of mania and depression, with a lifetime prevalence of 0.7%–6% [1–3].
Given its high morbidity, disability, and premature mortality [4], BD is a major health problem with severe socioeconomic implications. The economic burden of BD has been estimated at $45 billion per year in the United States in 1991, with a total of $7 billion of direct costs consisting of expenditures for inpatient and outpatient care [5, 6]. A very recent report analyzing the inpatient care costs for bipolar I disorder in Europe demonstrated that huge resources and frequent hospitalizations are required, especially in managing manic episodes [7]. Moreover, it has been shown that BD has a significant negative effect on work relationships, attendance, and functioning, with an overall increase in costs deriving from lost productivity [8]. Family, twin and adoption studies have demonstrated that BD is characterized by a high heritability, suggesting that genes exert a high-magnitude effect on disease development [9, 10]. In light of this evidence, great effort has been made to identify the genetic factors involved in the pathogenesis of BD, although fulfilling results have not yet been obtained. Investigations carried out to date on a genome-wide level using case-control study designs have thrown light on two genes, the ankirin G (ANK3) and the alpha 1C subunit of the L-type voltage-gated calcium channel (CACNA1C) [11].
Consequently, a number of studies have further investigated the role of these genes in BD. In regard to ANK3, its association has been displayed ex novo or replicated in several other studies, although with different effect size and association magnitude for the polymorphisms analyzed [12–16]. Following the first report of its association with BD [17], CACNA1C has been further investigated in some studies. In 2008, using the Wellcome Trust Case-Control Consortium dataset as comparison [18], a genome-wide association study (GWAS) of BD identified concordant association signals in the CACNA1C gene, although the genome-wide significance was not reached [13]. Since then and upholding the report by Ferreira et al. [11], the possible causative role of CACNA1C in BD has been confirmed by gene-wide analysis [19], although further evidence is needed to assess its effects on the disease [20]. Very recently, a meta-analysis of genome wide association data on major mood disorders, including BD, has identified a risk locus on 3p21.1 with the most significant association signal located in the protein polybromo-1 (PBRM1) and guanine nucleotide binding protein-like 3 (GNL3) genes, whose encoded products are, respectively, involved in chromatin remodelling and the proliferation of stem cells [21]. Among the genes possibly contributing to BD susceptibiliy the diacylglycerol kinase eta (DGKH), encoding for an enzyme that phosphorylates the second messenger diacylglycerol, was first found to be associated with BD in the GWAS by Baum et al. [12], although replication was not uniformly displayed by subsequent studies [22–24].
On the whole, this evidence seems to indicate that the biological underpinnings of BD appear to be highly heterogeneous on the genetic and molecular level. Moreover, it is unlikely that common variants of large effect-size are causative of BD, if, for instance, meta-analysis of GWAS data in BD displayed relatively low odds ratios (OR) of 1.45 and 1.18 and minor allele frequencies (MAF) in cases of 0.07 and 0.38 for ANK3 and CACNA1A genes, respectively [11].
This highly heterogeneous genetic framework characterizing BD reflects on the broad variability of its phenotypic expression, with clinical symptoms presenting different patterns within subjects (longitudinal variability) and between subjects [25–27].
The genetic and phenotypic heterogeneity of BD clearly plays a pivotal role in pacing the way to the discovery of biological determinants of the disease. Specifically, indisputable associations between the genetic variants identified to date and the phenotypic changes manifested in BD are still lacking. In this context, an appropriate phenotype definition represents a powerful tool in the search for susceptibility genes in BD [27, 28]. To this end, several subphenotypes have been studied to refine the phenotype and consequently increase the possibility of detecting genetic determinants [27, 29]. The response to long-term treatment with mood stabilizers has been suggested to be a clinical trait suitable for identifying distinct subgroups of BD with greater homogeneity and for the mapping of genes involved in the biological basis of treatment response and in the disorder itself [27, 30, 31].
On the one hand, the reduction of heterogeneity might depend on the identification of common phenotypic patterns in the clinical presentation of the disease [27]. On the other hand, the narrower are the criteria for the definition of treatment response, the greater the probability of reducing genetic heterogeneity and consequently of detecting causative determinants of differences in response to medications.
Phenotypic assessment is the harbinger of more complex research strategies such as pharmacogenomics, which aim to assemble the molecular and phenotypic heterogeneity jigsaw puzzle in the study of treatment response. Indeed, pharmacogenomics is the genome-wide analysis of the role of genetic determinants in modulating an individual's response to a drug treatment or the onset of adverse drug reactions (ADRs). Along with its use as a strategy for reducing heterogeneity and consequently for shedding light on the biological underpinnings of the disease, the integration of pharmacogenomic data with specific individual phenotypic information, represents a powerful instrument for the development of personalized therapies.
Although the data collected by numerous studies have not yet reached a sufficient magnitude to significantly impact the clinical field, pharmacogenomics in BD is a rapid-evolving research field. Specifically, most of the pharmacogenomic studies performed in BD have focused on candidate genes selected according to the biological rationale supporting their involvement in the drugs’ mechanism of action, in the biological basis of the disease and in the metabolic pathways implicated in the onset of ADRs.
In this paper, we present the current state of the pharmacogenomic research on long-term treatments in BD, specifically focusing on mood stabilizers. In the introductory section, we discuss the issues pertaining to the currently-available mood stabilizing treatments for BD, the appropriate definition of the treatment response phenotype and its implication for pharmacogenomics studies. Then we selectively review existing evidence on genetic predictors of maintenance treatment response in BD, firstly presenting an overview of the molecular targets involved in mood stabilizers mechanism of action and then focusing on presently available reports on the genetic determinants of treatment response.
2. Pharmacogenomics in Bipolar Disorder: Phenotypic Features and Heritability Patterns of Long-Term Treatment Response
2.1. Long-Term Treatments in Bipolar Disorder: Lithium, Anticonvulsants, and Antipsychotics
It is widely accepted that the term “mood stabilizers”, although not yet uniformly validated, may be applied to several classes of drugs, such as anticonvulsants, antipsychotics (particularly the second generation antipsychotics or atypical), and lithium which, although different in terms of mechanism of action and chemical structure, commonly act as prophylactic agents able to prevent illness recurrences. Ideally, a mood stabilizer should be efficacious in: (1) treatment of acute manic symptoms, (2) treatment of acute depressive symptoms, (3) prevention of manic symptoms, and (4) prevention of depressive symptoms [32]. Among the mood stabilizing agents, lithium, more than the others, seems to meet these criteria, further strengthening the rationale for its use as first-line treatment in BD [32–35]. In this regard, numerous controlled and open trials have clearly shown that lithium is able to reduce the frequency of episode recurrences by at least 30% [33, 35]. A randomised open-label trial recently demonstrated that both combination therapy with lithium plus valproate (VPA) and lithium monotherapy are more likely to prevent relapse than is valproate monotherapy [36]. Therefore, it is not surprising that since its first introduction in the clinical use in 1949 by Cade [37], lithium has remained one of the mainstays in the treatment of BD, a practice further corroborated by its high efficacy in reducing suicidality in patients with recurrent major affective disorders [38, 39].
Along with lithium, anticonvulsants and antipsychotics, represent valid alternatives for the therapeutic management of BD. Among anticonvulsants, three drugs [VPA, lamotrigine (LTG) and carbamazepine (CBZ)] have strong evidence-based support for use in clinical states of BD [40, 41]. Specifically, VPA, LTG and CBZ have data supporting their roles as potential long-term treatments to prevent relapse [40, 41]. LTG has a greater effect on the prevention of depression while has lacked efficacy in mania or in acute bipolar depression [41]. On the other hand, VPA is effective in the treatment of mania, somewhat more effective in certain patient subgroups (mixed mania, mania with prominent irritability) than other treatments; recent proof-of-concept indicates benefits in bipolar depression, while there is clear evidence that CBZ is effective in treating mania [41, 42]. Among the other anticonvulsants, gabapentin (GBP) and topiramate (TPM) have not been shown to be superior to placebo in randomized trials, although GBP might be useful in controlling anxiety in BD [41].
Antipsychotics, and specifically the class of atypical, appear to be effective in the treatment of mania, although their potential in the long-term management of BD is still subject of debate [43, 44]. A systematic review of randomized trials for treatment of the maintenance phase in BD supported evidence that, along with lithium, LTG and VPA, olanzapine is effective in long-term use significantly reducing manic relapses [45].
Although this evidence describes a background of multiple therapeutic tools available for the treatment of BD, a relatively high percentage of patients still present lack of response even after consecutive trials of different drugs. In this context, it appears crucial to develop theoretical frameworks useful in identifying clinical predictors of response by categorizing BD patients according to different diagnostic spectra. In this regard, a three-spectrum model of BD has been proposed, describing different subtypes of BD (classical, psychotic and characterological) characterized by specific clinical features and more importantly by specific patterns of drug treatment response [25]. This model supports the importance of the careful diagnostic assessment of BD at the phenomenological level, as some specific clinical features, such as family history, clinical course and comorbidity are strictly linked to the treatment response phenotype.
2.2. The Phenotype First
Definition of the treatment response phenotype is a crucial process propaedeutic to any subsequent pharmacogenetic/genomic analysis the value and reliability of the latter based on an accurate assessment of the former. Diagnoses are expected to be heterogeneous in BD, and similarly, quantification of the treatment response in clinical terms, a complex trait in itself, may vary. In detail, evaluation of treatment response outcomes has for the most part been performed considering it a binary-trait (0 = unaffected/nonresponders, 1 = affected/responders). However, it has been suggested that, since treatment response is a quantitative trait, this approach might not totally represent the complex nature of this phenotype. Indeed, deeming treatment response a quantitative trait can facilitate the identification of genetic variants involved in intermediate phenotypes, as for instance partial response to a drug. It is important to note that the binary-trait approach does not take into account the degree of variation in the treatment response phenotype, which can be represented as a gradient lying within the two extremes of complete nonresponse and complete response. On the other hand, comparing and contrasting the genetic differences at the two extreme points of the gradient (extreme discordant phenotype (EDP)) can increase the statistical power and consequently the probability of detecting the causative variants for the response or lack of response to a drug [46, 47].
In BD, efforts thus far to develop a stringent definition of treatment response, especially in long-term therapies, have applied analytical tools such as the Affective Morbidity Index (AMI) [48, 49] and the illness severity index [50]. Briefly, these approaches take into consideration the severity and the duration of episodes before and after the introduction of the mood stabilizing therapy, allowing quantification of the degree of improvement under a specific treatment. However, these approaches do not take into account the presence of possible confounders (such as additional medication or lack of compliance) that may interfere in the establishment of a causal relationship between the clinical improvement and the treatment. In this regard, in 2002 Grof et al. [51] developed a rating scale that measures the degree of improvement in the course of treatment and weighs clinical factors considered relevant for determining whether or not the observed improvement is due to the treatment. This scale allows enucleating the full response phenotype, meaning the absence of abnormal mood episodes during treatment. Moreover, since the scale consists of eleven items (0–10), it also permits identification of intermediate response phenotypes (i.e., partial response) and the EDP approach, both aspects supporting its validity for the implementation in pharmacogenomic studies of treatment response.
In general, the assessment of treatment response phenotype in BD shows quite similar rates of reduction of illness recurrence among the different mood stabilizing therapies. In regard to lithium, clinical data show that ∼80% of chronically-treated subjects are at least partial responders and that ∼30% of these patients are excellent responders with a complete remission of symptoms [52]. In a review of 20 randomized, double-blind controlled studies of monotherapy or combination therapy for the treatment of acute mania, the pooled response rates of 15 monotherapy studies were around 50% for lithium, VPA, CBZ or atypical antipsychotics (21% greater than placebo), while the remaining 5 combination studies showed an incremental benefit of adding a second agent equal to 21% [53].
However, treatment refractoriness in BD still remains a challenge [54], further encouraging the search for response predictors that might allow identification of the most effective treatment for carriers of specific genetic-variation patterns.
2.3. The Phenotypic Features and Heritability Patterns of Long-Term Treatment Response
In light of the complexity of the drug response phenotype, the pharmacogenomic approach might benefit from analysis of convergent findings on prediction of treatment response on the phenotypic and genetic level. It has been suggested that pharmacogenomic studies should consider nongenetic factors that can interact in influencing the phenotype [55]. Genetic work can be informed and guided by appropriate clinical details and can allow investigation of possible genotype-phenotype information. In BD, this joint analytical approach has led to intriguing findings on significant interactions between lithium treatment response, gene variation and specific clinical features [56].
Since clinical markers of treatment response can have predictive power that may allow the targeting of therapies to subpopulation of patients characterized by specific phenotypic traits, it is important to establish their correlation with treatment response outcome. In this context, a large amount of evidence has been gathered over the years on outcome predictors for long-term treatments with CBZ, VPA, LTG and lithium [57–59].
Treatment response to CBZ seems to be predicted by clinical features such as mood-incongruent psychosis, lack of response or intolerance to lithium, while contrasting findings are present on the possible predictive value of rapid cycling, severe mania and dysphoric mania [25, 57, 60]. Responders to VPA are characterized by the presence of pure, mixed or dysphoric mania. Moreover, early age at onset, rapid-cycling, concurrent substance abuse and lack of response or intolerance to lithium are clinical markers of positive outcome with VPA [57]. Response to LTG seems to be predicted by earlier onset of symptoms, non episodic course of illness, rapid cycling, comorbidity with panic or substance use disorder, fewer hospitalizations, fewer prior medication trials and male gender [61, 62].
Responders to lithium are a group of patients with distinct clinical features corresponding to the bipolar disorder core phenotype [33, 63]. An episodic pattern of mania-depression interval, low rates of comorbid conditions, absence of rapid cycling and high age at illness onset have been identified as potentially protective against a recurrence under lithium [59, 64–66].
In addition, family history might also provide important insights on the prediction of treatment outcome. For instance, relatives of LTG responders present a greater prevalence of schizoaffective disorders, major depression and panic attacks, while lithium responders had a higher risk of BD [59]. In this regard, it is important to note that since the first report by Mendlewicz et al. [67], which pointed out a significant correlation between response to lithium and the presence of BD in the proband's first degree relatives, this finding has been confirmed over a period of more than thirty years [50, 62, 68–74]. Moreover, Grof et al. [51] showed that lithium response in itself clusters in families. Analyzing the genetic patterns of heritability, Alda et al. [75, 76] suggested that a recessive model with sex-specific penetrance could explain the mode of inheritance in BD responsive to lithium.
On the whole, these findings clearly show that treatment response is a heritable clinical feature, evidence that corroborates the rationale for pharmacogenomic approaches in BD. Moreover, they underscore the importance of an accurate collection of clinical data which, if analyzed jointly with genetic information, could lead to precise prediction of treatment outcome in BD patients.
3. Molecular Mechanisms of Mood Stabilizers
Considerable evidence has shown that BD is characterized by neurotrophic alterations resulting in impairment of signalling and neuroplasticity with a significant overall reduction in central nervous system (CNS) volume (for a review, see [77]). Neuroimaging studies have demonstrated that BD and major depressive disorder (MDD) patients have several brain volumetric alterations such as enlargement of ventricles, reduced grey matter volumes in the orbital and medial prefrontal cortex (PFC) and reduced volumes in the frontal lobe and hippocampus [78–82]. Post-mortem studies have also shown that the reduced volumes of PFC and orbitofrontal cortices are associated with decreased neuron numbers and/or size [83, 84]. Other studies reported reductions in the number of interneurons in the anterior cingulated cortex of BD patients as compared to controls as well as reductions in nonpyramidal neurons in the hippocampus [85, 86].
It has been shown that the clinical efficacy of lithium correlates with measurable changes in the CNS of treated subjects, who displayed increased volumes in specific brain areas after chronic treatment. Recently, a longitudinal study conducted from 1997 until 2004 evaluated the effect of lithium on grey matter volumes of different brain areas of 28 bipolar subjects with different response to lithium medication, using morphometric magnetic resonance imaging [87]. A significant increase in total brain grey matter volume was identified after chronic lithium administration. Interestingly, the regional analysis showed that the grey matter volume of PFC was significantly increased only in lithium responsive BD subjects. These data show that the effect of lithium on the CNS has clinical consequences and corroborate the evidence on its neurotrophic effect. Nevertheless, the mechanism by which lithium increases grey matter volumes is still under investigation.
A large body of evidence shows that lithium interacts with many elements of second messenger systems thus modulating processes both up- and downstream of its targets. Lithium competes with magnesium for a large number of enzymes requiring it as a cofactor [88, 89]. Early reports show that lithium directly inhibits two enzymes of the inositol pathway, the inositol monophosphate phosphatase (IMPase) and the inositol polyphosphate 1 phosphatase (IPPase), at therapeutic serum concentration (0.6–1.2 mM) [90]. This evidence led to the inositol depletion hypothesis of lithium's mechanism of action, which would exert its mood stabilizing effects by decreasing inositol levels, thus ultimately influencing a number of processes relying on this pathway, including neurotrophin and G protein mediated signalling [91]. This hypothesis is also supported by findings from animal studies [92–94].
Like lithium, VPA has been shown to have neuroprotective effects. It has been reported that VPA interacts with the inositol pathway, but through a different mechanism compared to lithium. VPA does not directly inhibit IMPase, and the inositol depletion is mediated by a decrease in inositol phosphates and inositol levels [95, 96].
A number of findings have also shown that lithium and VPA interact with the activator protein 1 (AP-1), a family of transcription factors composed of heterodimeric complexes of c-Jun and c-Fos proteins. Lithium has been shown to increase AP-1 binding activity and transcription in cultured cells [97, 98]. Other studies have reported that VPA increases AP-1 DNA binding and activation in cultured cells [99, 100]. However, while lithium regulates AP-1 through the inhibition of glycogen synthase kinase 3β (GSK-3β), VPA induces the expression of c-Jun and c-Fos by means of histone deacetylase (HDAC).
GSK-3β is a highly-conserved protein involved in the regulation of apoptosis, circadian rhythm and a wide range of neuronal functions and pathways involved in cell and tissue development (for a review, see [101]). A large number of processes and pathways converge on GSK-3β including the Wingless-INT (Wnt) signalling, several neurotransmitter pathways, and Akt, with the latter shown to inhibit GSK-3β in response to multiple hormones and neurotrophic factors, including the brain derived neurotrophic factor (BDNF) [102].
Lithium, like VPA, inhibits GSK-3β activity in mouse and rat brain at therapeutically relevant doses [103–105]. It has been reported that lithium inhibition of GSK-3β is mediated through competition for magnesium, an essential cofactor for GSK-3β, while VPA inhibition is indirect [89, 106, 107]. The inhibitory effect of lithium and VPA on GSK-3β may represent one of the key mechanisms through which they regulate neuronal plasticity and neurogenesis. In this regard, it has been shown that alternative inhibitors of GSK-3β affect neurogenesis in embryonic stem cells [108–110] and stabilize growth cones in cultured sensory neurons [111]. Along with lithium, VPA and CBZ were shown to inhibit the collapse of sensory neuron growth cones and to increase growth cone area [112].
It has been shown that the serotonergic pathway is a downstream target of GSK-3β [113]. This evidence led to the hypothesis of an involvement of GSK-3β in depression and might explain the effectiveness of lithium as an add-on in the treatment of antidepressant-refractory depression [114, 115]. In this regard, a recent finding showed that lithium decrease serotonin release in primary serotonergic neurons from rat raphe nuclei [116]. In the same study, the authors reported that short-term lithium treatment (8 days) resulted in a 45% decrease in tryptophan hydroxylase 2 (TPH2) expression and a 31% reduction in TPH2 protein levels, completely compensated after long-term treatment (14 days).
As we mentioned earlier, the Wnt signalling pathway converges on GSK-3β. The activation of this pathway leads to the stabilization and then accumulation of β-catenin and is involved in gene expression regulation [117]. Lithium was shown to stabilize β-catenin through inhibition of GSK-3β, while VPA interacts with the Wnt pathway by increasing β-catenin transcription.
Among the elements interacting with GSK-3β and involved in neurogenesis, the family of cAMP response element binding proteins (CREB) is one of the most widely investigated. Lithium antagonizes phosphorylated CREB loss and upregulates both CREB and BDNF in several brain regions [118]. Moreover, BDNF levels in the rat brain [119–121] are increased after treatment with either lithium or VPA. A recent finding from a gene expression study performed on cultured rat cortical neurons showed that the treatment with lithium or VPA induced activation of the promoter IV of BDNF and that this activation was mediated by the inhibition of GSK-3 and HDAC, respectively [122]. HDAC is a key enzyme responsible for the hypoacetylation of histone proteins, resulting in gene silencing [123, 124], thus modulating transcription of about 2% of transcribed genes [125]. As a whole, these data show that VPA can potentially modulate a large number of long term processes through the epigenetic regulation of gene expression.
An effect of lithium on BDNF was also shown by Tseng and coworkers in lymphoblasts from BD patients responsive to lithium [126]. The authors reported that lithium treatment in vitro decreased BDNF levels more significantly in BD patients than in controls.
Another key element in neuroprotection is the B-cell CLL/lymphoma 2 (bcl-2), a protein involved in regulating cell survival by blocking apoptotic death. It has been shown that administration of both lithium and VPA induces a two-fold increase in bcl-2 levels in the frontal cortex of chronically-treated rodents [127] and in the neuronal cell lines SH-SY5Y [128].
As discussed in Section 2, newer anticonvulsants, such as LTG, TPM and GBP, are effective in the maintenance treatment of BD (LTG) or as add-ons to first line mood stabilizers (TPM, GBP). In contrast to lithium and VPA, less is known about the mechanism of action of these drugs. A number of studies have shown that LTG inhibits glutamate release through sodium and calcium channel blocking [129–131]. Moreover, LTG significantly enhanced the surface expression of GluR1/2 AMPA receptor in a time- and dose-dependent manner in cultured hippocampal neurons [132]. LTG was also shown to affect GABA-A receptor-regulated functions in the CNS. A gene expression study performed on primary cultured rat hippocampus cells treated with LTG 0.1 mM showed that the expression of the GABA-A receptor beta3 subunit was increased by the treatment [133]. LTG was also shown to inhibit GSK-3β in human neuroblastoma cells, a mechanism that is shared with VPA and lithium [134].
TPM is an antagonist of the glutamate receptors KA/AMPA. In addition to this function, TPM was shown to inhibit glutamate release in epileptic rats [135]. TPM not only affects glutamate, but also regulates GABA neurotransmission. Several studies have shown that TPM increased brain GABA levels in healthy humans [136] and enhanced GABA-stimulated Cl− influx into cerebellar granule neurons and cerebral cortical neurons [137]. Numerous findings also suggest that, like other mood stabilizers, LTG and TPM play a key role in neuroprotection. LTG has been shown to have neuroprotective effects in animal models of ischemia [138–141] and in the neuronal damage induced by excitotoxins, mitochondrial toxins and axotomy [142–144]. As regards GBP, the mechanism of its psychotropic action is still unknown, but some findings show that it has neuroprotective effects in ischemic animal models [145]. Moreover, some evidence suggests that GBP could negatively regulate glutamate neurotransmission [146].
3.1. Microarray Gene Expression Studies in Humans
The first report from a genome wide gene expression (GWGE) study on human derived cell lines was published in 2008 from Seelan and coworkers [147]. In this study, human neuroblastoma cells were maintained in therapeutic levels of lithium for 33 days. The transcriptome analysis showed that a total of 671 genes were differentially regulated after lithium treatment. Altered transcripts included genes of the apoptotic system and the phosphoinositide metabolism. In another study, Plant and colleagues [148] performed a transcriptome analysis on human neuroblastoma cells (SH-SY5Y) treated with either lithium or valproic for 6 h or 72 h. Findings showed that the treatment with both drugs determined an altered expression of 936 genes with the homeodomain protein Six1 being the most significantly upregulated. Moreover, the evaluation of the antiapoptotic action of lithium and valproate showed that Six1 over-expression protected the cells from staurosporine-induced apoptosis via the blockade of caspase-3 activation.
Another GWGE study was carried out on multiple prostate human cancer cell lines that were administered with therapeutic levels of lithium [149]. Lithium significantly inhibited cell proliferation, which was associated with reduced DNA replication and S-phase cell cycle arrest. Moreover, lithium significantly decreased the expression of multiple DNA replication-related genes.
During the 8th Annual Pharmacogenetics in Psychiatry Meeting [150], Mclnnis reported on a gene expression study performed on lymphoblasts from BD subjects, incubated with 1mM lithium chloride for 4, 8 and 16 days. Results revealed that the chromosome 8 open reading frame 33 (C8orf33) was significantly over-expressed after lithium treatment and that a total of 217 genes was significantly downregulated. C8orf33 activity was found to be associated with G protein-coupled receptor protein signalling pathway, neuroactive ligand-receptor interaction, Ca++ signalling pathway and the regulation of the actin cytoskeleton.
Recently, Sugawara and colleagues [151] evaluated the effect of both lithium and valproate on gene expression levels in lymphoblasts derived from three healthy subjects. The microarray analysis revealed that 44 and 416 genes were regulated by lithium and valproate, respectively. Among the 18 genes commonly altered, the strongest downregulation was reported for the vascular endothelial growth factor A (VEGFA) gene. As for lithium-specific effects, the most significant altered expressions were reported for the BCL-2 associated X protein (BAX) and the platelet-activating factor acetylhydrolase isoform 1b, beta subunit (PAFAH1B2).
3.2. Microarray Gene Expression Studies in Rodents
In 2002, Bosetti and coworkers carried out a GWGE to describe gene expression changes in brain of rats fed with lithium for 7 (subacute) and 42 (chronic) days respectively using an array of 4132 genes [152]. Findings showed that lithium downregulated 25 genes after 7 days but did not upregulate any gene. In contrast, after 42 days of lithium treatment, 50 genes were upregulated while no gene was downregulated. Interestingly, among the downregulated genes was the inositol polyphosphate-1-phosphatase gene (INPP1), whose expression was 2.7 times lower after 7 days and 2.6 times lower after 42 days of lithium treatment.
Another microarray gene expression study was performed by McQuillin and coworkers in 2007 [153]. Findings showed that lithium caused a significant change in the expression level of 121 genes. Among these, lithium upregulated the period homolog 2 (Per2), the secretogranin II (Scg2), the BDNF and the IMPA1 genes. Other interesting findings consisted of the upregulation mediated by lithium of the deiodinase (DIO2) and thyroid hormone receptor interactor 12 (TRIP12) genes, encoding for elements that might be involved in the development of hypothyroidism, a long-term side effect caused by lithium treatment in clinical settings.
Another study by Chetcuti et al. [154] identified a significantly-differential expression of a number of genes in brains of mice treated with lithium for 7 days as compared to a control sample. Validation by quantitative PCR showed that five genes were differentially expressed after lithium treatment. These included genes involved in metal ion homeostasis and chemical/electrical gradients across membranes, and regulating RNA polymerase II, protein degradation and G-protein-coupled signal transduction.
As regards to VPA, a recent study examined gene expression levels in rat cortical neurons treated with VPA using microarray technology and reported that 726 genes were upregulated while 577 were downregulated by the treatment. Interestingly, the expression of BDNF was upregulated by VPA while the expression of the α4 subunit of the GABA-A receptor gene (GABA-ARα4) and the K+/Cl− cotransporter (KCC2), which are both involved in the development of GABAergic inhibitory neurons, were downregulated [155].
3.3. MicroRNAs Studies on Lithium Mechanism of Action
MicroRNAs (miRNA) are a class of endogenous post-transcriptional regulators binding to complementary sequences in the 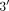 UTR of multiple target mRNAs. The miRNA database (miRBase: http://www.mirbase.org/) currently include more than 10000 entries but this number will presumably increase rapidly in the coming years. A single
miRNA can regulate the expression of a large number of genes, usually resulting in their silencing. Among their functions,
miRNAs have been shown to regulate neurite outgrowth, neurogenesis and synaptic plasticity, mainly through the regulation
of mediators such as CREB, methyl CpG binding protein 2 (MECP2), the fragile X mental retardation 1 protein (FMRP) and elements
of the Wnt pathway [156].
UTR of multiple target mRNAs. The miRNA database (miRBase: http://www.mirbase.org/) currently include more than 10000 entries but this number will presumably increase rapidly in the coming years. A single
miRNA can regulate the expression of a large number of genes, usually resulting in their silencing. Among their functions,
miRNAs have been shown to regulate neurite outgrowth, neurogenesis and synaptic plasticity, mainly through the regulation
of mediators such as CREB, methyl CpG binding protein 2 (MECP2), the fragile X mental retardation 1 protein (FMRP) and elements
of the Wnt pathway [156].
A recent study explored the role of miRNAs in the mechanism of action of lithium in a sample of lymphoblasts from BD patients [157]. Each cell line was divided into two lines: one line was treated with lithium 1 mM for 4, 8 and 16 days while the other was grown in lithium-free medium. Results showed that lithium treatment determined expression changes in 7 miRNAs after 4 days treatment and that 4 out of these 7 were significantly regulated after 16 days. The most intriguing finding was reported for miR-221 and miR-34a, two miRNAs previously shown to be regulated by lithium and VPA in rat hippocampus [158]. These miRNAs are respectively responsible for regulating the expression of 29 and 10 genes, including some genes previously shown to be implicated in BD. Interestingly, the study by Zhou and colleagues also showed that in primary culture of hippocampal neurons treated with lithium or VPA, levels of the protein glutamate receptor metabotropic 7 (GRM7), one of the miR-34a targets, was increased thus showing that miR-34a might contribute to mediating the effect of lithium and VPA on GRM7 [158].
4. Pharmacogenomics of Mood Stabilizers
4.1. Linkage Studies on Lithium Response
As a result of its efficacy and wide clinical use, pharmacogenomic studies on BD have for the most part focused on response to lithium prophylaxis. Early linkage studies used pharmacogenomic approaches based on lithium response as a strategy for reducing heterogeneity in BD. In 1999, Ewald and colleagues carried out a haplotype sharing analysis on chromosome 18 in a sample of eight lithium responding BD subjects from the Faroe Islands who had common ancestors [159]. The study reported increased haplotype sharing on the distal part of chromosome 18 (region 18q23). In 1999, Turecki and coworkers reported a modest linkage with markers in the chromosome region 18q22.3 in unilateral families of BD patients responsive to lithium [160]. Interestingly, the same region was previously shown to be associated with bipolar in a sample of narrowly defined BDI patients for which, however, the response to mood stabilizing treatment was not evaluated [161].
Another linkage study on BD probands for which response to lithium treatment was considered as inclusion criteria, [162] reported significant LOD scores for the region of chromosome 12q23-q24 in a large pedigree from the homogeneous population from Saguenay-Lac-St-Jean (Quebec). More recently, Turecki and colleagues [163] performed a genome scan on 31 families of probands with excellent response to lithium using 378 markers. The most significant linkage was found with markers in chromosome regions 15q14 and 7q11.2. However, when the response phenotype was considered in the analysis, the 15q14 region appeared to be more probably involved in BD phenotype and 7q11.2 in lithium response.
4.2. Candidate Gene Studies on Lithium Response
As shown by the large amount of evidence on the key role of the inositol pathway in the mechanism of action of lithium, numerous candidate gene studies on lithium response have been performed on inositol-related genes. However, while some authors reported intriguing findings, a strong evidence of high impact genes is still lacking.
The gene encoding for phospholipase cω1 (PLCG1), a key enzyme involved in G protein mediated signals and in the inositol pathway, has been investigated in several studies. One paper reported association for a dinucleotide repeat in PLCG1 in a sample of 136 BD patients excellent responders to lithium and 163 controls [167]. The same study reported a modest linkage with this polymorphism in a sample of unilineal families. However, these findings were not replicated in a re-examination study performed in a sample of Norwegian lithium-treated bipolar patients sub-classied as lithium responders, nonresponders, or partial responders/unclassied [174]. PLCG1 was further investigated in the study by Ftouhi-Paquin and coworkers [175], in which the authors screened the gene for functional polymorphisms. While three single nucleotide polymorphism (SNPs) in the translated region of PLCG1 were identified, none of the markers was found to be associated with BD in a sample of 133 excellent responders to lithium and 99 healthy controls.
Other genes encoding for elements of the inositol pathway have been tested for association with lithium response. The INPP1 was first investigated by Steen and coworkers in 1998 [166]. The authors showed association for the silent variant C937A and response to lithium in a Norwegian sample of 23 BD patients and 20 controls but not in a sample of 54 BD and 50 controls from Israel. Nevertheless, a recent study failed to confirm the association for this polymorphism and lithium response in a sample of 134 BD patients comprised of 61 full responders, 49 nonresponders and 24 partial responders to lithium [186]. Positive association with lithium response was also shown for two polymorphisms in the gene encoding the inositol monophosphatase 2 (IMPA2) in the study by Dimitrova and colleagues [183]. However, the relatively small size and power of the BD sample characterized for lithium response did not allow the authors to draw definitive conclusions.
The gene encoding for the IMPA1 has been studied in two papers reporting no association with lithium response [56, 165]. Several studies have investigated for association another gene encoding for a key element in lithium mechanism of action: the GSK-3β gene [56, 184–186]. However, among the four studies performed so far, only Benedetti and coworkers reported association between the −50 T/C polymorphism and lithium response, thus providing no conclusive results [184].
Another set of interesting findings was produced for the BDNF gene. In 2005, Rybakowski and colleagues [181] reported positive association between SNP rs6265 (Val66Met) and better response to lithium in a sample of 88 BD patients. The Val66Met is a functional polymorphism with the Met allele shown to be associated with lower depolarization-induced secretion of the protein in neurons [195]. The Val/66Met polymorphism was investigated by Rybakowski and coworkers in a further study reporting an interactive effect between the Val/Val genotype and the s/s and l/s genotypes of the serotonin-transporter-linked promoter region (5-HTTLPR) polymorphism in a sample of 107 BD subjects characterized for lithium response [196]. Other association studies of 5-HTTLPR polymorphism in lithium response are discussed hereafter in this review.
Another study by the same group recently investigated for association four SNPs in the BDNF gene and three SNPs in the gene encoding for the neurotrophic tyrosine kinase, receptor, type 2 (NTRK2) in a sample of 108 BD patients. Findings showed association between the BDNF Val66Met polymorphism and degree of response to lithium, but no association was reported for NTRK2 polymorphisms [188]. The same gene was also investigated in the paper by Bremer et al. [56] in which significant association between lithium response and two SNPs within NTRK2 was shown in BD patients with suicidal ideation as well as in BD patients with posttraumatic stress disorder. Interestingly, in 2009 Szczepankiewicz and coworkers [191] have investigated the association between the Src-family tyrosine kinases (FYN) gene and lithium response in a sample of 101 BD patients characterized for lithium response. FYN is a member of the protein-tyrosine kinase oncogene family and belongs to the protein kinase family phosphorylating NMDA receptor subunits, participating in the regulation of ion transmission and BDNF/TrkB signal transduction pathway. In this study the authors reported a trend for association for one of the polymorphisms with a worse response to lithium. While the study does not strongly support the involvement of this gene in lithium response, findings are intriguing in the light of a previous paper by the same group in which an association for FYN polymorphisms and BD was reported [197]. As a whole, these results corroborate the evidence indicating a key role of the BDNF/Trk signaling pathway in lithium response and BD.
Another interesting finding was reported in the paper by Mamdani and coworkers, in which the authors investigated the association between CREB1, CREB2 and CREB3 genes and response to lithium in a sample comprised of 180 BD lithium responders, 69 non responders and 127 controls [189]. Findings showed association between CREB1-1H and 7H SNPs and response to lithium.
As we mentioned earlier, several authors showed that the DGKH gene was associated with BD. Squassina et al. [22] recently investigated the three SNPs previously found to be associated with BD by Baum et al. [12], in a sample of 197 Sardinian BD I patients, 91 of whom were characterized for lithium response. While the authors failed to replicate the association for individual SNPs, a significant association with BD was reported for a haplotype of the three SNPs. However, the study did not show statistically significant association with response to lithium. Moreover, this lack of association has been confirmed in an extended cohort of Sardinian BD patients charachterized for lithium treatment [198].
Along with inositol pathway genes, a number of papers have also dealt with genes encoding for elements of neurotransmitters’ systems. The 5-HTTLPR polymorphism has been investigated in lithium response by a number of authors. Three studies reported association between the short allele (s) and/or genotypes carrying the s allele and a worse response to lithium [176, 178, 179]. Another study investigated the interactive effect of 5-HTTLPR and BDNF Val66Met in a sample comprised of 31 patients classied as excellent responders, 54 as partial responders and 26 as nonresponders to lithium prophylaxis [187]. Findings showed that patients with 5-HTTLPR s/s and l/s genotypes having the BDNF Val/Val genotype were more frequent in non responders than in the other groups.
While to date the association with 5-HTTLPR has been replicated most frequently in lithium response studies, other authors failed to report positive findings for this polymorphism [186, 194].
Another interesting finding for serotonin-related genes was reported in a paper by Serretti et al. [169] in which the authors identified an association between variants in the TPH gene and worse response to lithium in a sample of 90 BD and 18 major depressive patients.
A number of studies have also dealt with dopamine system genes, but to date only one paper showed positive association. Rybakowski and coworkers reported an association between the G allele and G/G genotype of the −48 A/G polymorphism in the DRD1 gene and worse response to lithium [190]. A detailed list of pharmacogenetic studies in lithium response can be found in Table 1.
Pharmacogenetic studies in lithium response.
4.3. Genome Wide Association Studies on Lithium Response
To date only one GWAS has been published reporting findings on a sample of bipolar patients characterized for lithium response [202]. The results showed that a SNP in the glutamate receptor ionotropic, AMPA 2 (GLUR2) gene was associated with time to recurrence. However, the association did not reach the genome wide significance and the characterization of lithium response of the replication sample was performed using a different methodology.
Some international joint efforts are being undertaken to identify the genetic variants involved in the modulation of lithium response. The Consortium on Lithium Genetics (ConLiGen, www.conligen.org), created in 2008, has already collected a sample of 1,500 BD patients retrospectively assessed using narrow criteria for the phenotypic characterization of treatment response [203]. The GWAS analysis is currently ongoing, and new genetic findings on lithium response and related ADRs will soon be available to the scientific community.
4.4. Gene Expression Studies on Lithium Response
Only a small number of gene expression studies have been performed thus far using human tissues from patients characterized
for the response to lithium. In 2004, Sun and coworkers [204] published findings from a microarray gene expression study on lymphoblasts from BD patients excellent responders to lithium.
After chronic lithium treatment in-vitro, the authors found that the expression levels were altered for 7 genes, 5 of which
were confirmed by Northern blotting analysis. These 5 genes codified for elements of pathways involved in neurotransmission
and in processes that might be related to the lithium mechanism of action, such as the alpha I B-adrenoceptor (αIB-AR), acetylcholine
receptor protein alpha chain precursor (ACHR) and cAMP-dependent 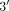 ,
, 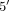 -cyclic phosphodiesterase 4D (PDE4D).
-cyclic phosphodiesterase 4D (PDE4D).
A recent study measured the expression levels of the gene codifying for the PDZ and LIM domain 5 (PDLIM5) protein in a sample of BD patients characterized for lithium response and a sample of controls [193]. PDLIM5 is an adaptor protein that selectively binds the protein kinase C epsilon (PKC∊) to its target, the N-type Ca2+ channels, voltage-gated Ca2+ channels specically expressed in axons [205]. The study failed to show significant differences in expression levels between lithium responders and partial/non responders or controls.
4.5. Pharmacogenomic Studies on other Mood Stabilizers
Compared with lithium, much less evidence is available on the pharmacogenomics of other mood stabilizing treatments. A recent study investigated the association between the −116C/G polymorphism in the X-box binding protein 1 (XBP1) gene and reponse to VPA in a sample of 51 bipolar patients [198]. XBP1 is a an endoplasmic reticulum (ER) stress-responsive transcription factor regulating major histocompatibility complex (MHC) class II genes by binding to a promoter [206]. The study showed an association between the G allele (that is associated with reduced transcription activity of XBP1) and a better response to VPA as compared to the C allele. The XBP1 gene was also shown to be involved in the pathophogenesis of BD [207], thus supporting the notion that its product might play a role in both the disease and the reponse to mood stabilizers.
Another candidate gene study examined the role of the Val158Met polymorphism in the catechol-O-methyl transferase (COMT) gene in response to mood stabilizers in a sample of 144 BDI patients and 157 controls [201]. During the treatment period, all patients were under mood stabilizer therapy with either lithium, VPA or CBZ. The results showed that the Met/Met genotype was more frequent in nonresponders than in responders, while no differences were detected between BD patients and controls. However, since the sample was not sub-divided according to the different types of treatment, it is not possible to draw conclusions on the role of COMT Val66Met in the response to specific mood stabilizers.
To our knowledge, only one pharmacogenetic study based on response to LTG has been performed. More precisely, Perlis and coworkers examinated the association between 19 SNPs located in several genes and response to Olanzapine/Fluoxetine combination (OFC) or LTG in a sample of 88 OFC-treated and 85 LTG treated bipolar I depression patients [200]. In regard to LTG treatment, findings showed that SNPs within the dopamine D2 receptor (DRD2), dopamine β-hydroxylase (DBH), glucocorticoid receptor (NR3C1), histamine H1 receptor (HRH1) and melanocortin 2 receptor (MCR2) genes were associated with the treatment response. Table 2 lists most of the association studies carried out to date in response to other mood stabilizing medications.
Pharmacogenetic studies in response to other mood stabilizing medications.
4.6. Pharmacogenomic of Antipsychotics in Bipolar Disorder
Despite the constant increase in the use of atypical antipsychotics in the maintenance treatment of BD, still very few studies have investigated the genetic underpinnings of their therapeutic response. In 2006, Dévila et al. [199] investigated the role of COMT Val158Met polymorphism on psychotic features in 42 BDI patients, further testing the hypothesis of a possible influence of this variant on blood plasma concentration of metabolites of dopamine and noradrenaline and on the severity of the disorder and the response to olanzapine and lithium. No significant findings emerged with regard to treatment response to olanzapine as well as for the other markers tested. Aiming to evaluate common genetic variation for association with clinical improvement in BDI depression following treatment with OFC, Perlis et al. [200] analyzed a cohort of 88 patients treated with OFC, showing significant association for dopamine D3 receptor (DRD3) and HRH1 genes and response to OFC. The dearth of pharmacogenomic studies on antipsychotics in BD should encourage research efforts aiming to identify possible genetic predictors of response, especially in light of the ever-wider use of these agents as maintenance treatments.
5. Summary and Future Perspectives
Pharmacogenomic approaches in BD have thus far concentrated on identifying genetic predictors of treatment response to mood stabilizers. However, based on the evidence that lithium responsive BD patients share common clinical features, pharmacogenomics has also been used as strategy for enucleating subgroups of individuals characterized by lower phenotypic and presumably genetic heterogeneity, thus also possibly increasing the power of detecting causative variants of BD. While linkage studies performed on BD patients responsive to lithium have highlighted some chromosomal regions (7q11.2, 15q14, 18q23), further studies have reported no association for specific genes within these regions. Candidate-gene approaches have so far focused on genes codifying for elements of biological pathways shown to be target of lithium, such as proteins of the intracellular second messenger cascade mediated by inositol, Wnt and neurotrophins pathways and the GSK-3β protein. Along with findings from gene expression studies, these data have provided intriguing, although yet inconclusive, insights into the understanding of the genetic underpinnings of lithium response.
As regards to the anticonvulsant VPA, the small number of pharmacogenetic studies performed so far failed to provide evidence for high impact genes. However, the associations reported for XBP1 and COMT genes are still intriguing in light of their key role in the mechanism of action of VPA and in pathways putatively involved in modulating the pathophysiology of BD. As for LTG, evidence for association with treatment response has thus far been reported for DRD2, β-hydroxylase, glucocorticoid receptor, HRH1 and melanocortin 2 genes. While these findings come from the only candidate gene study performed to date, they shed light on genes encoding for elements shown to be involved in both the LTG mechanism of action and BD.
Overall, genetic data on response to mood stabilizers are informative tools that along with data on phenotypic and environmental factors might be useful in predicting individual treatment response profiles thus paving the way to targeted therapies.
To facilitate the implementation process from pharmacogenomics to personalized medicine in the long-term treatment of BD, various research areas still need to be developed.
For instance, findings are still substantially lacking as regards safety pharmacogenomics, an approach dedicated to avoiding ADRs [208]. Phenotypically, an ADR from a drug is generally assessed more objectively. Such events usually occur a short time after the patient experiences the drug, so the precise phenotypic response can be specifically, sensitively and accurately documented [208]. Since it could accurately define individuals who might be at higher personal risk of an adverse event, safety pharmacogenomics acquires particular importance in mood stabilizing treatments in BD, given their long-term nature and their specific pharmacokinetic/pharmacodynamic properties. For instance, lithium is characterized by a narrow therapeutic index, and its clinical management is greatly impacted by important side effects, above all the alteration of thyroid function and nephrogenic diabetes insipidus. Moreover, the other available therapeutic tools, anticonvulsants and antipsychotics, also present a wide range of side effects ranging from allergic reaction to haematological toxicity and weight gain. In clinical practice, these side effects can dramatically affect the positive outcomes of long-term treatments, since patients having a high degree of response may be forced to stop effective maintenance treatments due to the onset of serious ADRs.
In conclusion, once pharmacogenomics produces clear data to reliably determine the extent of the genetic contribution to the treatment response phenotype and the predictive power of positive response outcome associated with them, the personalized therapy strategy can be quite easily integrated into clinical settings ultimately leading to the selection of drugs that will be both safe and effective for a patient.
Financial Disclosures
The authors of this paper do not have any commercial associations that might pose a conflict of interest in connection with this paper.
Acknowledgments
The authors gratefully thank Ms. Mary Groeneweg for commenting on the paper. This work was partly supported by the Regional Councillorship of Health, “Regione Autonoma della Sardegna” with a grant dedicated to Drug Information and surveillance Projects to A. Squassina, M. Manchia, M. Del Zompo. A. Squassina is supported by a Grant from “Regione Autonoma della Sardegna”, and the European Social Fund (ESF), LR7 2007. A. Squassina and M. Manchia contributed equally to the paper.
- Received March 15, 2010.
- Accepted June 24, 2010.
- © 2010 Alessio Squassina et al.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- Amari L.,
- Layden B.,
- Rong Q.,
- Geraldes C. F. G. C.,
- Mota De Freitas D.
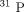 NMR, and
NMR, and 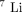 NMR spectroscopic methods for investigating Li+/Mg2+ competition for biomolecules”. Analytical Biochemistry, vol. 272 no. 1 pp. 1–7 1999 2-s2.0-0033564972 doi:10.1006/abio.1999.4169.
NMR spectroscopic methods for investigating Li+/Mg2+ competition for biomolecules”. Analytical Biochemistry, vol. 272 no. 1 pp. 1–7 1999 2-s2.0-0033564972 doi:10.1006/abio.1999.4169. - ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵















