TABLEAU CLINIQUE DU PATIENT
DR BOYLE : Monsieur, merci d'être venu aujourd'hui. Pouvez-vous nous
dire quelques mots sur vous?
MR Y : Merci de me recevoir. J'ai 52 ans, j'ai grandi en Caroline du Nord.
Je suis maintenant marié et je vis en Virginie où j'ai
travaillé comme ingénieur pendant les 23 dernières
années. Et j'ai une mucoviscidose.
DR BOYLE : Pouvez-vous nous parler de la manière dont le diagnostic
a été fait.
MR Y : Et bien, je pense que mes parents suspectaient que quelque chose
n'allait pas peu de temps après ma naissance car j'avais constamment
d'importantes selles graisseuses. Mais ce n'est que lorsque j'ai eu une
pneumonie à l'âge de 3 ans que j'ai eu un test de la sueur et que
le diagnostic de mucoviscidose a été établi.
DR BOYLE : Savez-vous ce qui a été dit à vos parents
sur votre futur état de santé?
MR Y : On leur a dit de ne pas trop s'attacher à moi car j'avais peu
de chances de vivre plus de 7 ans.
DR BOYLE : Et lorsque vous avez atteint l'âge de 7 ans?
MR Y : On leur a dit que je pourrais atteindre 10 ans.
DR BOYLE: Racontez-nous votre enfance. Avez-vous été souvent
malade?
MR Y : Non, en dehors de prendre des enzymes pancréatiques et
d'avoir une désobstruction des voies respiratoires, je me sentais un
enfant normal. J'avais des infections sinusales récidivantes, qui
nécessitaient des antibiotiques, mais j'étais capable de faire
la même chose que les autres enfants et était même assez
athlétique. En fait, au collège, je pouvais courir un mile (1609
m) en 5 minutes.
DR BOYLE : Quel type de problèmes de santé avez-vous eu plus
tard?
MR Y : Plus tard au collège, j'ai développé une
pneumonie et après cela, j'ai eu une toux grasse assez
régulièrement. Mais je n'ai jamais eu besoin d'un traitement
intraveineux par antibiotiques et je ne me sentais pas limité par ma
maladie. Au cours de mes années 20 et 30, j'agissais pour de nombreuses
choses comme si je n'avais pas de mucoviscidose. Mes soins consistaient en une
consultation occasionnelle avec mon interniste local, en la prise d'enzymes
pancréatiques et dans l'inhalation d'albutérol si
nécessaire. Ceci a changé toutefois lorsque j'ai atteint mes
quarante ans. J'ai commence à avoir une aggravation de l'essoufflement
et des infections pulmonaires plus sévères. J'ai aussi
développé une sinusite sévère, qui a
nécessité une chirurgie. En raison de l'essoufflement qui
s'aggravait et des infections fréquentes, j'ai décidé de
trouver un centre spécialisé dans les soins des patients adultes
avec mucoviscidose.
|
|

|
|
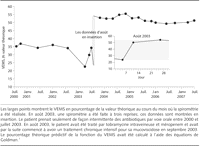
Figure 1.. Volume expiratoire maximal en une seconde (VEMS) de Mr Y entre 2000 to
2007
|
|
|
DR BOYLE : Je crois que lorsque vous êtes venu nous voir pour la
première fois au Johns Hopkins Adult Cystic Fibrosis Program, vous
n'aviez jamais eu de traitement prolongé par antibiotiques
intraveineux, ou tiré bénéfice d'un des nouveaux
traitements de la mucoviscidose actuellement disponible. Comment vous
êtes-vous senti après un traitement plus intensif?
MR Y : Avant le traitement, j'avais un sévère essoufflement,
même au repos, pour la première fois dans ma vie. Ceci
entraînait une anxiété relativement importante.
Après le premier traitement intraveineux antibiotique, Je me suis senti
une personne différente. J'étais capable de monter à
nouveau les escaliers et ne pas passer la majeure partie de ma journée
obnubilé par ma respiration et ma toux. Mieux, cette
amélioration a duré. Peu de temps après avoir
terminé les antibiotiques intraveineux, j'ai commencé à
prendre régulièrement de l'azithromycine, à inhaler de la
dornase alfa, de la tobramycine, et du sérum hypertonique, en
même temps qu'avec une désobstruction quotidienne intensive des
voies aériennes. Et je me suis senti incroy-ablement bien. Ce protocole
demande un investissement significatif chaque jour mais cela en vaut la
peine.
DR BOYLE : Ceci est agréable à entendre. Je pense que ce
graphique de votre fonction pulmonaire au cours des 7 dernières
années démontre clairement le bénéfice de tout
votre dur travail (FIGURE 1).
Le cas de Mr Y est instructif car il souligne plusieurs aspects importants de
la mucoviscidose: l'importance croissante des soins chez l'adulte, la
variabilité de la sévérité de la maladie et
l'efficacité des nouveaux traitements.
PATHOPHYSIOLOGIE
La mucoviscidose est une maladie multi-systémique qui entraîne
une maladie sino-pulmonaire chronique significative, une altération de
la fonction exocrine pancréatique, une élévation du
chlore dans la sueur, et une infertilité masculine. Toutes ces
anomalies phénotypiques sont provoquées par une dysfonction de
la protéine CFTR (cystic fibrosis transmembrane conductance
regulator).2
La protéine CFTR est un canal chlorique présent sur les
épithéliums de la plupart des lumières de l'organisme et
est un facteur contributif significatif à l'équilibre sodique et
hydrique. Le gène CFTR est situé sur le
chromosome7
et la mucoviscidose est provoquée par des mutations du gène qui
conduisent à un dysfonctionnement du canal
protéique.2
Plus de 1500 mutations ont été identifiées pouvant
conduire à un dysfonctionnement de la protéine CFTR et
entraînant un phénotype de type
mucoviscidose.3
Le mécanisme précis par lequel une dysfonction de CFTR
entraîne un phénotype de type mucoviscidose est inconnu, bien
qu'il soit clair que l'effet de la dysfonction de la protéine CFTR sur
les cellules épithéliales et glandulaires des voies
respiratoires conduit à une diminution de la profondeur de la couche
liquide de surface des voies respiratoires pulmonaires, une augmentation de la
viscosité des sécrétions des voies respiratoires et une
diminution de la capacité à contrôler les infections
bactériennes.4
La mucoviscidose exhibe un profil génétique classique de
transmission récessive, demandant deux 2 mutations pour être
présent pour que la maladie se développe. Les porteurs d'une
simple mutation n'ont généralement pas d'anomalies
phénotypiques. Ceci est heureux car environ 4% des tous les blancs sont
porteurs d'une mutation simple, ce qui fait de la mucoviscidose la maladie
autosomique récessive abrégeant la vie la plus commune au sein
de la population blanche (1 sur 3200 naissances
vivantes).5,6
La mucoviscidose est moins fréquente chez les non blancs, avec une
incidence d'environ 1 sur 15 000 naissance vivantes chez les noirs, 1 sur 9200
chez les latino-américains et 1 sur 10 000 chez les
asiatiques.6,7
Actuellement, il y a plus de 0 000 personnes atteintes de mucoviscidose aux
Etats-Unis et on estime qu'il existe environ 100 000 cas dans le
monde.8,9
|
|
|
|
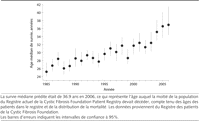
Figure 2.. Age de survie médian prédit dans la mucoviscidose entre 1985
et 2006
|
|
|
DIAGNOSIS
Le diagnostic de mucoviscidose demande un tableau compatible avec le
phénotype de mucoviscidose et la preuve biologique de dysfonction de
CFTR.10 Une
déclaration de consensus sur le diagnostic de mucoviscidose a
résumé les aspects clés du
phénotype.10
Ceux-ci incluent (1) une maladie chronique sino-pulmonaire se manifestant par
une toux chronique et une production de sputum, une infection persistante par
des pathogènes typiques de la mucoviscidose dont le Staphylococcus
aureus, l'Haemophilus influenzae, le Pseudomonas aeruginosa, et la
Burkholderia cepacia, une preuve radiographique de dilatation de bronche, et
une sinusite chronique, allant souvent avec une polypose nasale; (2) des
anomalies du tractus gastro-intestinal et nutritionnelles se manifestant par
une insuffisance pancréatique ou une pancréatite
récidivante, un ileus méconial ou un syndrome d'obstruction
intestinale distale, une impossibilité à bien se
développer ou une malnutrition chronique et des preuves de cirrhose
biliaire focale; et (3) des problèmes masculins uro-génitaux se
manifestant par une absence bilatérale congénitale des canaux
déférents et une azoospermie obstructive. Les personnes qui ont
un ou plusieurs de ces caractéristiques répondent aux
critères de phénotype de la mucoviscidose.
Mais beaucoup de ces traits n'étant pas spécifique, il est
essential qu'une preuve biologique de la dysfonction de CFTR soit
également établie avant de faire un diagnostic de mucovicidose.
Une dysfonction de CFTR peut être démontrée par une des
trois façons suivantes: (1) un test de la sueur avec un chlore
supérieur à 60 mEq/l (pour convertir en mmol/l, multiplier par
1.0); (2) un génotypage de CFTR démontrant la présence de
deux mutations connues de la mucoviscidose; ou (3) des anomalies
bioélectriques caractéristiques par mesure directe de la
fonction CFTR dans l'épithélium nasal. En raison de
l'association de la sensibilité (98%), de la spécificité
(95%), et des faibles coûts ($150-$300), le test de la sueur reste le
test de dépistage initial recommandé pour la mucoviscidose chez
les patients
suspects.11,12
Mais, d'un autre côté, 2% des personnes atteintes de
mucoviscidose n'ont pas de test de la sueur positif à 60 mEq/l ou plus
de chlore, ce qui oblige à démontrer la dysfonction de la CFTR
chez les patients pour lesquels il existe une forte suspicion clinique de
mucoviscidose et qui ont test de la sueur négatif.
La deuxième option est de rechercher la présence de deux
mutations pour la mucoviscidose; toutefois, la sensibilité du
génotypage de la mucoviscidose dépend totalement du nombre de
mutations testées et du contexte ethnique de la personne
testée.11
Environ 1500 mutations différentes ayant été
identifiées comme pouvant induire une mucoviscidose, la
sensibilité des tests de sueur dépasse le génotypage
à moins qu'une mutation extrêmement approfondie ne soit faite.
Ces dépistages de mutation approfondis sont maintenant disponibles dans
le commerce et comprennent le séquençage de tous les exons CFTR
et les séquences clés intron. Des considérations de
coût empêchent le génotypage d'être la
première ligne de dépistage des personnes ayant un
phénotype suggestif. Toutefois, un génotypage approfondi peut
être extrêmement valable chez les personnes ayant un test de la
sueur peu
concluant.13
Par ailleurs, des mesures directes in vivo de la fonction CFTR dans
l'épithélium nasal des personnes (différence nasal
potentielle) est disponible dans certains centres de mucoviscidose pour aider
au diagnostic dans les cas
difficiles.10
EPIDEMIOLOGIE
Au cours des 4 dernières décennies, la survie moyenne dans la
mucoviscidose a augmenté considérablement. On disait aux parents
d'enfants ayant une mucoviscidose, nés dans les années 1950s ou
1960s, comme les parents de Mr Y, qu'il était improbable que leurs
enfants dépassent leurs années d'adolescence. Avec le
développement des antibiotiques contre le pseudomonas et des
remplacements d'enzyme pancréatique, la survie a commencé
à devenir significative. Les développements continus des
traitements ont entraîné des améliorations presque
annuelles de la survie, permettant une survie médiane actuelle dans la
mucoviscidose de 36.9 ans (FIGURE
2).8
|
|
|
|
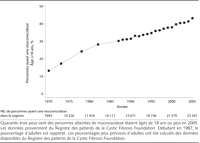
Figure 3.. Pourcentage d'adultes chez les personnes ayant une mucoviscidose
|
|
|
Cette augmentation de la survie a permis une augmentation
considérable du nombre d'adultes atteints de mucoviscidose
(FIGURE
3).8,14
Le nombre de personnes ayant une mucoviscidose, de 18 ans ou plus, s'est accru
de plus de 400% depuis le début des années 1970s, et
actuellement 43% de toutes les personnes ayant une mucoviscidose sont
âgées de 18 ans ou plus. Au cours de la prochaine
décennie, 50% de toutes les personnes atteintes de mucoviscidose auront
18 ans ou plus et la mucoviscidose deviendra une pathologie observée
plus souvent en médecine de l'adulte qu'en pédiatrie. Tandis que
la majorité des adultes atteints de mucoviscidose ont entre 18 et 30
ans, il existe un nombre croissant d'adultes âgés atteints de
mucoviscidose, avec 25% entre les âges de 30 et 39 ans, et 10% de plus
de 40 ans.8
L'année dernière, il y avait 10 individus de plus de 70 ans dans
le Registre des US Cystic Fibrosis Foundation National
Data.8
Bien que la raison la plus importante pour le personnel de soins de
connaître la mucoviscidose soit le nombre croissant d'adultes atteints
de mucoviscidose, il existe aussi une importance diagnostique potentielle. La
majorité des personnes atteintes de mucoviscidose est
diagnostiquée avant l'âge de 3 ans. Toutefois, environ 5% des
personnes atteintes de mucoviscidose sont diagnostiquées après
l'âge de 16
ans.8 Un
grand nombre de ceux-ci n'ont pas les symptômes classiques de la
mucoviscidose, posant des dilemmes potentiels pour les internistes qui ne
pensent pas toujours à la mucoviscidose lors du diagnostic
différentiel chez les adultes ayant une dilatation de bronches, une
sinusite chronique, une pancréatite récidivante, ou une
infertilité masculine. L'arrivée récente d'un
dépistage chez les nouveau-nés dans de nombreux états
diminuera probablement la fréquence des diagnostics tardifs de
mucoviscidose dans un futur
distant.15
TABLEAU CLINIQUE ET PRISE EN CHARGE
Tableau pulmonaire
La pathologie pulmonaire de référence observée dans la
mucoviscidose est l'obstruction bronchique muqueuse, l'inflammation et
éventuellement la dilatation de bronches. Le plus souvent, les
dilatations de bronches se présentent initialement dans les lobes
supérieurs puis progressent vers les lobes inférieurs avec le
temps. Lors de l'évaluation de la maladie pulmonaire, de nombreuses
études ont montré que le volume expiratoire forcé en une
seconde (VEMS1) est un bon facteur prédictif de mortalité chez
les individus ayant une
mucoviscidose.16,17
Tandis que des études antérieures avaient suggéré
qu'un VEMS inférieur à 30% de la valeur théorique
était associé à un taux de mortalité à 2
ans de 50%, de récentes données ont suggéré que
les résultats dans ce groupe se sont améliorés et il
existe une large variabilité dans l'expérience des patients
à l'échelle individuelle selon la fréquence des
poussées infectieuses et des comorbidités
associées.17-20
Malheureusement, il y a une perte progressive de la fonction pulmonaire
dans la mucoviscidose, qui commence dans l'adolescence et correspondant
à une perte moyenne de 1% à 4% par an. Toutefois, des taux
différents de déclin de la fonction pulmonaire donnent un
spectre assez large de sévérité de la maladie pulmonaire
chez les adultes atteints de mucoviscidose, environ 25% ayant une maladie
pulmonaire sévère, 40% modérée et 35%
légère ou ayant une fonction pulmonaire normale (sur les
critères du
VEMS).8 Les
personnes atteintes de mucoviscidose ont souvent une flore bactérienne
caractéristique dans leur sputum, avec une infection chronique par S
aureus et H influenzae chez les enfants et P aeruginosa chez les adultes.
Environ 80% des personnes atteintes de mucoviscidose sont chroniquement
infectées par P aeruginosa à l'âge de 18
ans.8
L'évolution clinique de la maladie pulmonaire dans la mucoviscidose
est marquée par des poussées périodiques,
caractérisées par une augmentation du sputum, de la
dyspnée, de la toux, de la fatigue, de la perte de poids, et une
diminution des valeurs
spirométriques.14
Ces exacerbations peuvent contraster avec les pneumonies classiques en cela
qu'elles ne sont pas habituellement accompagnées de fièvre,
d'infiltrats considérables à la radiographie pulmonaire, ou
d'hémocultures positives.
Prise en charge pulmonaire
Le traitement de la maladie pulmonaire dans la mucoviscidose comprend une
prise en charge à la fois aiguë et chronique. Les personnes ayant
des poussées aiguës sont traitées avec une
désobstruction intensive des voies respiratoires et des antibiotiques
en fonction des cultures bactériennes du sputum. La grande
majorité des adultes atteints de mucoviscidose étant
infectés par un P aeruginosa, 2 antibiotiques intraveineux contre le
pseudomonas sont typiquement administrés pendant 14 à 21
jours.14,21
Ces antibiotiques sont combinés avec des techniques de
désobstruction des voies respiratoires 3 à 4 fois par jour pour
aider au drainage des sécrétions infectées. L'objectif de
ce traitement est à la fois d'améliorer les symptômes et
de redonner à la personne un VEMS proche des valeurs de départ.
C'est ans le traitement de l'atteinte pulmonaire de la mucoviscidose qu'il y a
eu le plus de
progrès(Encadré).21
Ces traitements ciblent l'obstruction chronique, l'infection et l'inflammation
présentes dans les voies respiratoires.
L'obstruction est traitée d'abord par des techniques de drainage
mécanique, qui aident à défaire et drainer les
sécrétions visqueuses caractéristiques de la
mucoviscidose. Certaines de ces méthodes comprennent des oscillations
à haute fréquence de la paroi thoracique avec un gilet, des
oscillations des voies respiratoires hautes avec une valve flottante, et des
percussions thoraciques
manuelles.22
Tout ceci sert à défaire les sécrétions et est
suivi par des épisodes de respiration profonde destinée à
éclaircir les voies respiratoires. L'augmentation du nombre des
viscosités dans les sécrétions pulmonaires des patients
atteints de mucoviscidose étant due à l'ADN des neutrophiles
impliqués dans l'infection chronique pulmonaire, de la DNase
recombinante humaine (dornase alfa) est souvent inhalée pour diminuer
la viscosité du sputum. L'utilisation quotidienne de dornase alfa
inhalée a montré dans plusieurs études qu'elle
améliorait le VEMS et diminuait la fréquence des poussées
aiguës
pulmonaires.23
Récemment, l'inhalation de sérum salé hypertonique
à 7% a aussi démontré son efficacité dans le
drainage des voies respiratoires dans la
mucoviscidose.24,25
Avec 2 à 4 inhalations par jour, le sérum salé
améliore le VEMS et diminue la fréquence des exacerbations.
Dans le traitement de l'infection pulmonaire chronique de la mucoviscidose,
les antibiotiques inhalés sont utilises. Les antibiotiques par voie
orale en traitement chronique anti-pseudomonas sont rarement utilisés
en raison du risque de développement de résistance à la
seule classe orale d'antibiotiques actifs contre le pseudomonas, les
fluoroquinolones. Les antibiotiques inhalés, offrent toutefois un grand
nombre d'options antibiotiques avec l'avantage de donner de fortes
concentrations pulmonaires de médicaments avec une absorption
systémique minime. Inhalée deux fois par jour en traitement
chronique dans un protocole un mois sur deux, la tobramycine a montré
qu'elle améliorait significativement le VEMS et diminuait la
fréquence des poussées de
mucoviscidose.26
Bien que ceci puisse se produire, le développement de
résistances a été moins fréquent que ce que l'on
pensait initialement. D'autres antibiotiques inhalés qui ont
montré un bénéfice potentiel en utilisation chronique
incluent le colistiméthate et
l'aztréonam.21
Enfin, il existe un intérêt croissant pour les traitements qui
peuvent diminuer l'inflammation intrinsèque des voies respiratoires
dans la mucoviscidose. L'utilisation quotidienne chronique d'azithromycine par
voie orale a montré dans 4 études séparées qu'elle
pouvait diminuer la fréquence des exacerbations dans la
mucoviscidose.27
Le mécanisme de cet effet est encore sous investigation car on ne sait
pas s'il est dû à la réduction de l'inflammation ou aux
effets antimicrobiens. De nombreux autres essais explorant les traitements
anti-inflammatoires dans la mucoviscidose sont actuellement en cours.
| Encadré. Règles d'or du traitement de la
mucoviscidose
Drainage des voies respiratoires (percussion manuelles, gilet oscillatoire,
valve flottante)
Dornase alfa mucolytique (2.5 mg nébulisée
quotidiennement)
Antibiotiques nébulisés
Tobramycine (300 mg deux fois par jour un mois sur deux)
Colistiméthate, aztréonam
Azithromycine orale (500 mg/jour 3 fois par semaine)
Sérum salé hypertonique inhalé (4 ml à 7% 2-4
fois par jour)
Traitement antibiotique intensif lors des poussées
Soutien nutritionnel (régime riche en calories et en sel)
Remplacement des vitamines liposolubles A, D, E, K
Exercice
|
|
Ces traitements sont associés à un suivi régulier de
la spirométrie et à une intervention rapide avant un
déclin pulmonaire dans un effort pour minimiser la perte de fonction
pulmonaire. Les recommandations de soins chez l'adulte suggèrent aussi
l'utilisation d'imagerie intermittente radiologique pour aider à suivre
la progression de la maladie pulmonaire, tout en essayant de compenser pour la
nécessité d'une imagerie détaillée et l'exposition
aux
radiations.14
Alors que les traitements disponibles peuvent avoir un impact significatif
à la fois sur les symptômes et la fonction pulmonaire, il peuvent
demander 1 à 2 heures chaque jour, ce qui est un problème majeur
pour les adultes à l'école, au travail ou en famille. Les
recherches actuelles se focalisent sur des stratégies qui
améliorent la rapidité et l'efficacité de
l'administration des traitements de la
mucoviscidose.28
Si le déclin chronique des valeurs de la fonction pulmonaire
résulte en un VEMS constamment inférieur à 30% de la
valeur théorique, les personnes commencent souvent à ressentir
à la fois une limitation significative de leurs activités
quotidiennes et le besoin plus fréquent d'hospitalisation et
d'antibiotiques intraveineux. C'est souvent l'association d'une diminution du
VEMS et une augmentation des symptômes qui conduit les individus
à être adressés pour une évaluation initiale en vue
d'une double transplantation
pulmonaire.19
Le moment exact de cette transplantation pulmonaire dépend de la
sévérité ultérieure des symptômes et du taux
de déclin clinique car les études ont montré que seuls
les patients les plus atteints ont un bénéfice de survie
à 5 ans après
transplantation.29
Les personnes atteintes de mucoviscidose subissant une transplantation
pulmonaire font aussi bien ou mieux que les personnes ayant une pathologie
conduisant à une transplantation
pulmonaire.19
Environ 150 personnes avec mucoviscidose subissent une transplantation
pulmonaire chaque année, soit environ annuellement 1.5% de la
population adulte atteinte de mucoviscidose. Toutefois, il existe seulement un
taux de survie de 50% à 5 ans après cette procédure et
peu de preuves que l'avènement d'une transplantation ait modifié
la survie médiane globale estimée dans la
mucoviscidose.19,29
Une bactérie inhabituelle qu'il est bon de connaître car elle
peut influencer significativement l'évolution pulmonaire de la
mucoviscidose est la B cenocepacia, une espèce génomique
spécifique du complexe B cepacia qui est redoutée dans la
communauté de la mucoviscidose car elle peut entraîner un
déclin rapide de la fonction pulmonaire et est capable d'être
transmise entre les individus atteints de
mucoviscidose.30
Des poussées de B cenocepacia avec une morbidité et une
mortalité significatives ont poussé à faire des
recommandations de contrôle strict des infections dans les centres
spécialisés dans la mucoviscidose et concernant le rassemblement
des personnes atteintes de
mucoviscidose.31
Burkholderia cenocepacia est souvent résistante aux antibiotiques
standards anti-pseudomonas et peut nécessiter un traitement avec
plusieurs médicaments dont le méropenem et l'association
triméthoprim/sulfaméthoxazole.21
D'autres complications fréquentes qui peuvent rendre la prise en
charge de l'atteinte pulmonaire chez les adultes atteints de mucoviscidose
plus difficile sont l'infection à P aeruginosa résistant
à plusieurs antibiotiques
(17%-25%),32
l'infection à des mycobactéries non tuberculeuses (7%-13%),33 et
le développement d'une aspergillose allergique broncho-pulmonaire
(2%-7%).34
Questions gastro-intestinales
Bien que le facteur déterminant le plus puissant de la durée
et de la qualité de vie dans la mucoviscidose soit l'équilibre
pulmonaire, les questions nutritionnelles et gastro-intestinales jouent aussi
un rôle significatif. Environ 90% de personnes ayant une mucoviscidose
ont une insuffisance pancréatique exocrine et ont besoin d'utiliser une
supplémentation orale pancréatique avec les repas pour
prévenir une malabsorption
chronique.8
Cette pathologie pancréatique est souvent notée sur les
scanners sous la forme d'une « substitution graisseuse du
pancréas ». La plupart des patients atteints de mucoviscidose
prennent des suppléments pancréatiques contenant un
mélange d'amylase, de lipase, et de protéase avant chaque repas.
Même en utilisant des suppléments enzymatiques
pancréatiques, les personnes atteintes de mucoviscidose ont encore un
degré de malabsorption graisseuse. L'association d'une malabsorption
graisseuse et d'une maladie pulmonaire chronique peut entraîner des
besoins caloriques extrêmement élevés, demandant aux
personnes atteintes de se concentrer sur les repas riches en calories (de
même que sur des repas riches en sel pour compenser l'excès de
perte sodique dans la
transpiration).14,35
Des quantités supplémentaires de vitamines liposolubles A, D,
E, K sont aussi nécessaires. Souvent, en dépit des efforts pour
optimiser l'apport calorique, la malnutrition empêche d'atteindre la
croissance maximale et le poids
potentiel.35
Des options thérapeutiques dans ce contexte incluent l'utilisation de
stimulants de l'appétit ou la mise en place d'une sonde de gastrostomie
pour permettre une alimentation nocturne. La Cystic Fibrosis Foundation a
fixé comme objectif d'indice de masse corporelle 22 pour les femmes
atteints de mucoviscidose et 23 pour les hommes (calculé comme le poids
en kilogrammes divisé par la taille en mètres au
carré).36
Les études ont suggéré une forte association entre le
statut nutritionnel et le résultat, aussi des recommandations
récentes de prise en charge ont souligné l'importance de faire
très attention au statut
nutritionnel.14,20,36
Une autre manifestation fréquente de la pathologie du tractus
gastro-intestinal chez les adultes atteints de mucoviscidose est le syndrome
d'obstruction intestinale distale. Ceci est classiquement provoqué par
un épaississement des selles et du mucus dans l'iléum terminal
et se présente sous la forme d'un tableau clinique d'obstruction de
l'intestin
grêle.14
Le syndrome d'obstruction intestinale distale se développe souvent dans
le contexte d'une malabsorption chronique, une non observance à la
supplémentation enzymatique, d'une déglutition de mucus,
à une déshydratation ou à l'utilisation de
narcotiques.
L'obstruction répond habituellement au traitement avec une solution
orale de polyéthylène glycol et par des lavements avec produits
de contraste hydrosolubles. Une autre question potentielle gastro-intestinale
chez les adultes atteints de mucoviscidose inclut l'élévation
chronique des taux des enzymes hépatiques, une cirrhose avec
hypertension portale, et une augmentation du risque de lithiases
cholédocienne et
rénale.14
Bien que les mécanismes exacts soient inconnus, l'augmentation de la
viscosité des sécrétions et la malabsorption
gastro-intestinale caractéristiques de la mucoviscidose sont
considérées comme des facteurs contributifs majeurs de toutes
ces complications.
Insuffisance endocrinienne
Au moment où ils atteignent l'âge adulte, 20% à 30% des
personnes atteintes de mucoviscidose développent une insuffisance
pancréatique endocrine en plus de leur insuffisance exocrine, le
début typique de cette complication se situant entre les âges de
18 et 24
ans.37 Le
diabète lié à la mucoviscidose (CFRD) est un type unique
de diabète qui a des caractéristiques différentes
à la fois du diabète insulinodépendant et non
insulinodépendant. Le diabète lié à la
mucoviscidose est provoqué par la possibilité pour le
pancréas atteint de produire une petite quantité d'insuline,
mais insuffisante pour répondre complètement à l'apport
des hydrates de carbone. Ceci fait que le CFRD ne se complique presque jamais
d'acidocétose, mais est fréquemment caractérisé
par une hyperglycémie significative après les repas. Pour
éviter une perte de poids, le CFRD est rarement pris en charge par un
régime. A la place, un traitement est habituellement mis en place avec
de l'insuline à action brève au moment de repas.
Un diabète pauvrement contrôlé, qui peut
entraîner une dysfonction des neutrophiles et une malnutrition, est
associé à une accélération de la perte de la
fonction pulmonaire et une augmentation du risque de mortalité chez les
personnes atteintes de
mucoviscidose.37,38
Une prise en charge intensive du CFRD est donc devenu une partie importante
des soins de la
mucoviscidose.37
D'autres maladies endocrines fréquentes de la mucoviscidose chez
l'adulte incluent l'ostéopénie et l'ostéoporose, environ
les 2/3 des adultes atteints de mucoviscidose ayant une diminution du contenu
minéral
osseux.39
Alors que le déficit en vitamine D est extrêmement
fréquent chez les adultes atteints de mucoviscidose et contribue
souvent à une diminution de la densité osseuse, la diminution du
contenu minéral osseux observée dans la mucoviscidose est
probablement le résultat de facteurs multiples dont les cytokines de
l'inflammation chronique, des taux inadaptés de testostérone, la
malnutrition et l'effet direct des mutations CFTR sur le développement
osseux.39
Tissus reproductifs
Environ 99% des hommes atteints de mucoviscidose ont une absence
congénitale bilatérale de canaux déférents
(CBAVD), ce qui a les mêmes effets qu'un homme sain ayant eu une
vasectomie.14
Bien que le canal déférent soit anormal, les hommes atteints de
mucoviscidose produisent normalement du sperme et sont donc capables d'avoir
des enfants grâce à des techniques de reproduction
assistée comme l'aspiration du sperme micro-épididymale et
l'insémination in vitro. D'un autre côté, les femmes
atteintes de mucoviscidose ne semblent avoir de diminution significative de
leur capacité à féconder. Des études ont
démontré que les femmes atteintes de mucoviscidose ayant un
état pulmonaire et nutritionnel adéquat peuvent avoir une
grossesse
normale.40,41
En raison de l'hérédité récessive, les descendants
des personnes affectées par la mucoviscidose ne seront pas
affectées sauf si leur partenaire est porteur de la mutation de la
mucoviscidose (ou a aussi une mucoviscidose).
LA MUCOVISCIDOSE ET L'INTERNISTE
Le nombre croissant d'adultes atteints de mucoviscidose et le fait que 5%
d'entre eux sont diagnostiqués après l'âge de 16 ans rend
crucial le fait pour les praticiens de patients adultes de devenir
accoutumé à la mucoviscidose. Une chose dont il faut se souvenir
est qu'une dilatation de bronche plus une des trois choses suivantes doit fait
penser à la mucoviscidose: (1) infertilité masculine (5% des cas
d'infertilité masculine sont dus à une CBAVD et environ 80% des
CBAVD sont associés à des mutations de CFTR 42); (2) une
pancréatite idiopathique récidivante (10%-20% des personnes
ayant une pancréatite chronique idiopathique sont porteuses de
mutations de la mucoviscidose 43); ou (3) une polypose nasale
récidivante (une sinusite bactérienne récidivante avec
polypes du nez peut être un tableau clinique classique de formes
légères de mucoviscidose).
En conséquence, toute combinaison d'infertilité masculine, de
pancréatite non expliquée ou de polypes du nez
récidivants, en particulier dans le cadre d'une dilatation de bronches,
doit immédiatement conduire à une évaluation
complémentaire à la recherche d'une mucoviscidose. Les autres
diagnostics qui doivent être envisagés chez les personnes ayant
une maladie pulmonaire ressemblant à une mucoviscidose incluent la
dyskinésie ciliaire, les déficits en immunoglobulines, et
l'aspergillose allergique bronchique. L'évaluation initiale de ces
affections doit inclure une microscopie électronique de la structure
des cils et les taux sériques des IgG et des IgE.
RESEAU DE SOINS POUR LA MUCOVISCIDOSE DE L'ADULTE
En raison des besoins particuliers de soins des adultes atteints de
mucoviscidose, la Cystic Fibrosis Foundation a établi des
recommandations pour être accrédité comme centre de
référence de la mucoviscidose de l'adulte. Un centre doit avoir
un médecin ou plus ayant l'expertise de la mucoviscidose chez l'adulte
et une équipe multidisciplinaire spécialisée incluant une
infirmière coordinatrice, une diététicien, un
kinésithérapeute respiratoire, et un(e) assistant(e) social(e)
focalisés sur le soutien de l'adulte atteint de mucoviscidose. La liste
des centres accrédités est disponible à
http://www.cff.org.
SUIVI À 6 MOIS
Mr Y a continue à se consacrer à son protocole quotidien de
soins et à utiliser son passé d'ingénieur en suivant ses
progrès à l'aide d'un tableau. Au cours des trois
dernières années, il n'a pas eu besoin d'être
hospitalisé et n'a pas eu besoin de traitement antibiotique
intraveineux. Il dit que l'utilisation régulière du drainage des
voies respiratoires, l'inhalation d'antibiotiques et de mucolytiques lui a
« rendu sa vie ».