|
|
MALADIES NEUROLOGIQUES
La sclérose latérale amyotrophique
La sclérose latérale amyotrophique (SLA) ou maladie de
Charcot ou maladie de Lou Gehrig correspond à la perte progressive des
motoneurones (type de cellules nerveuses contrôlant les muscles) du
cortex cérébral et de Lmoelle épinière. C'est une
maladie progressive, invalidante, de cause inconnue, dont l'issue est
fatale.
Le déplacement, la parole, la déglutition, la respiration, et
autres fonctions de base sont affectés avec le temps. Environ 30 000
américains sont concernés par la SLA. L'incidence est de 1
à 2 cas sur 100 000 individus par an. Elle est
généralement décelée à l'âge
mûr et affecte plus d'hommes que de femmes. Le numéro du JAMA du
11 juillet présente un article sur le diagnostic de la SLA et les
recommandations en termes de soins palliatifs.
SYMPTOMES
On estime que plus de 50% des motoneurones sont perdus avant que les
symptômes, comme la faiblesse musculaire, deviennent apparents.
- Faiblesse musculaire progressive et fonte musculaire au niveau des bras et
des jambes
- Fasciculations (contractions involontaires apparentes des
muscles)
- Difficultés à déglutir, parler et respirer.
- Raideur musculaire, douleurs corporelles, crampes, surtout la nuit
La défaillance respiratoire est la cause la plus fréquente du
décès. La pneumonie figure parmi les autres causes. Dans plupart
des cas, le décès survient 3 à 6 ans après les
premiers symptômes, bien que certains individus affectés par la
SLA vivent plusieurs années de plus, voire plusieurs dizaines
d'années.
DIAGNOSTIC
Le diagnostic est basé sur une histoire médicale rigoureuse,
un examen physique et des analyses en laboratoire. Les
électromyogrammes (EMG), mesures de la conduction nerveuse pour
évaluer les fonctions neuromusculaires, constituent les analyses
clés. Les autres analyses peuvent inclure des analyses de sang et des
analyses de neuro-imagerie, comme une tomodensitométrie ou une IRM
cérébrale ou de la moelle épinière. Des analyses
moléculaires, des analyses de fluide cérébro-spinal ou
des biopsies musculaires peuvent être nécessaires.
TRAITEMENT
- Des thérapies physiques, occupationnelles ou de l'orthophonie
peuvent aider à la vie quotidienne
- La riluzole est le seul médicament reconnu pour le traitement de la
SLA. Elle peut prolonger la vie du patient de quelques mois.
- D'autres médicaments peuvent soulager les symptômes comme les
douleurs musculaires, crampes, salivation abondante, spasmes, et
asthénie.
SOINS PALLIATIFS
- Des exercices physiques adaptés pour aider au maintien des fonctions
motrices.
- Régimes destinés à minimiser les problèmes de
déglutition et d'assurer une alimentation correcte.
- L'utilisation d'appareillages comme les colliers cervicaux, orthèses
des membres inférieurs, cannes, déambulateurs, ou fauteuils
roulants.
- Rampes, mains courantes, siège de toilette surélevé,
siège de douche.
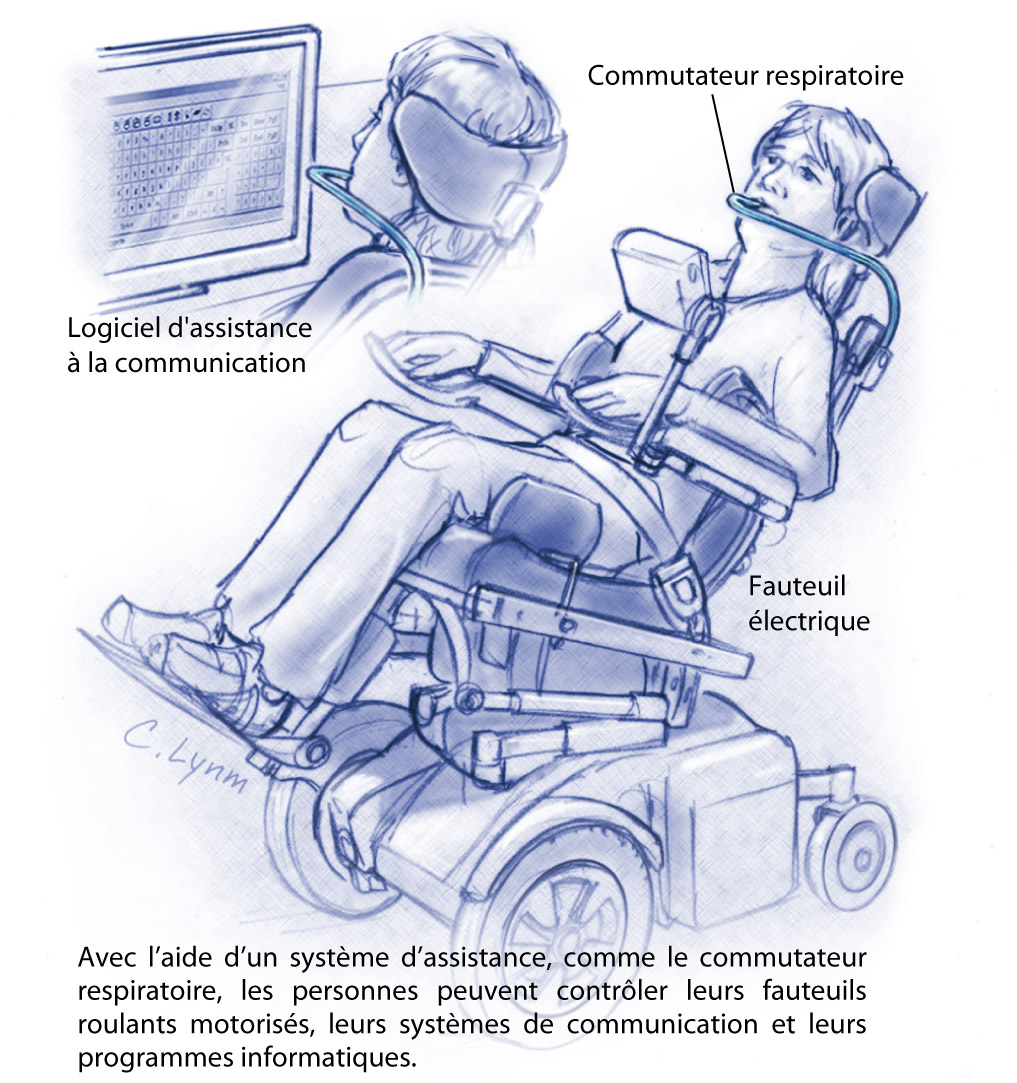
- Tablettes à écrire effaçables, amplificateurs vocaux
et ordinateurs pour faciliter la communication.
- Une ventilation non invasive (appareillage respiratoire) peut
s'avérer important pour assister à la respiration.
La nature progressivement invalidante de la SLA et le fait qu'il n'y ait
pas de traitement curatif en fait une maladie difficile à prendre en
charge. En plus des soins médicaux, le patient a besoin du soutien
psychologique de la famille, des amis, du médecin et des autres
personnels soignants.
POUR EN SAVOIR PLUS
INFORMEZ-VOUS
Pour retrouver cette page et les précédentes Pages du patient
du JAMA, visiter le lien Page du patient sur le site web du JAMA au
www.jama.com.
Beaucoup sont disponibles en espagnol et en
anglais.
Sources: Muscular Dystrophy Association, Amyotrophic Lateral Sclerosis
Association
La page du patient du JAMA est un service public du JAMA. Les informations
et les recommandations publiées dans cette page sont, dans la plupart
des cas, appropriées, mais elles ne se substituent pas au diagnostic
médical. Pour des renseignements spécifiques sur votre condition
médicale personnelle, le JAMA suggère que vous consultiez votre
médecin. Les médecins et autres professionnels de la
santé peuvent photocopier de manière non commerciale cette page
afin de la distribuer parmi leurs patients. Pour des réimpressions en
de nombreux exemplaires, veuillez composer le 203/259-8724.
John L. Zeller;
Cassio Lynm;
Richard M. Glass
ARTICLE EN RAPPORT
JAMA. 2007;298:139.
Texte Complet
|