|
|
PAGES DU PRATICIEN
Manifestation neurologiques de la maladie de von Hippel-Landau
John Butman, MD, PhD;
W. Marston Linehan, MD;
Russell R. Lonser, MD
JAMA. 2008;300(11):1334-1342
RÉSUMÉ
| |
La maladie de von Hippel-Lindau (VHL) est un syndrome de néoplasie autosomale dominante résultant d'une mutation de la lignée germinale du gène suppresseur de la tumeur de VHL sur le bras court du chromosome 3. Les patients souffrant de VHL présentent une prédisposition à développer des lésions du système nerveux central et des viscères. Les lésions du système nerveux central incluent des hémangioblastomes (tumeur la plus commune dans le cas de VHL) et tumeurs du sac de la poche endolymphatique (ELSTs). Les manifestations viscérales comprennent des carcinomes et des kystes rénaux, des tumeurs neuroendocriniennes et des kystes pancréatiques, des phéochromocytomes et des cystadénomes des organes reproducteurs annexes. En dépit de leur pathologie bénigne, les hémangioblastomes et les ELSTs sont une cause fréquente de morbidité et de mortalité chez les patients souffrant de VHL. Les récentes investigations en biologie moléculaire dans ces lésions du système nerveux central associées à la maladie de VH apportent une nouvelle perspective concernant leur origine et leur développement. Les données provenant de l’imagerie en série et de protocoles de surveillance clinique fournissent une perspective pour l'histoire naturelle de ces lésions. En raison de la pathobiologie dissemblable et de l’évolution clinique entre les hémangioblastomes et les ELSTs, les stratégies de gestion optimale de ces manifestations neurologiques de la maladie de VHL sont très différentes.
PRÉSENTATION DU CAS
| |
Cette femme âgée de 39 ans, a d’abord été examinée dans les Instituts Nationaux de Santé National Institutes of Health NIH) en août 2003. Trois ans avant son évaluation, elle a éprouvé un épisode aigu de vertige et de tintement de l'oreille gauche. Son IRM (Imagerie par Résonance Magnétique) du cerveau et des os temporaux étaient sans particularité, et un audiogramme a révélé une audition normale. Ses symptômes ont été attribués à une maladie de Ménière et elle a été placée sous un régime pauvre en sodium sans soulagement. Vingt-quatre mois plus tard, la patiente a éprouvé un épisode de perte soudaine de l'ouïe au niveau de l’oreille gauche avec exacerbation aiguë du vertige et du tintement de l’oreille. Un audiogramme a confirmé une perte auditive gauche neuro-sensorielle modérée à sévère. L’IRM a démontré une hémorragie dans le labyrinthe gauche mais pas de preuve de tumeur du sac endolymphatique (ELST). La patiente a continué son régime pauvre en sodium sans soulagement des signes ou symptômes audio vestibulaires.
Neuf mois après la perte auditive de la patiente, sa mère a été diagnostiquée cliniquement comme souffrant de la maladie de von Hippel-Lindau (VHL) après qu'il ait été trouvé qu’elle présentait des carcinomes bilatéraux rénaux (RCCs), des kystes pancréatiques, et des hémangioblastomes du Système Nerveux Central (CNS). Le diagnostic clinique a été confirmé par la recherche de la mutation de la lignée germinale de la maladie de VHL. Par la suite, la patiente a été confirmée comme présentant la maladie de VHL par critères cliniques et la présence du gène de la mutation de la lignée germinale de la maladie de VHL. La patiente a été confiée au NIH et lors de la semaine précédant l’évaluation, elle a éprouvé un début soudain de vertige et un tintement dans l'oreille gauche et en même temps une perte aiguë de l'ouïe à gauche (confirmée par audiométrie). Le scanner (CT) et l’IRM ont encore démontré une hémorragie intra labyrinthique gauche et une petite ELST dans l’aqueduc vestibulaire gauche. L’IRM de l’axe cranio-spinal a révélé des hémangioblastomes de l'obex médullaire et de la moelle épinière thoracique (Figure 1). Le scanner abdominal a révélé des RCCs bilatéraux, des kystes rénaux, et des kystes pancréatiques (Figure 2). Quatre semaines après l’évaluation, l'ELST a été réséquée, entraînant une résolution de son vertige et de son tintement d'oreille. Sa perte de l'ouïe est restée inchangée.
|
|
|
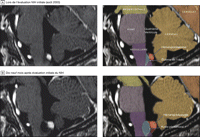
Figure 1. IRM Prise d'images de la résonance de l’hémangioblastome du tronc cérébral démontrant la formation de kyste Peritumoral
A révélé un hémangioblastome amplifiant de l'obex dans le medullaire et un œdème (hypointensité dans le tronc cérébral adjacent à la tumeur). La patiente était asymptomatique à ce moment. B, Dix-neuf mois après l'évaluation du NIH, la patiente présentait des maux de tête, hoquets fréquents et des difficultés pour avaler. L’IRM T1-pondérée en contraste renforcé a révélé le développement d'un kyste péritumoral associé à un hémangioblastome de l'obex.
|
|
|
|
|
|
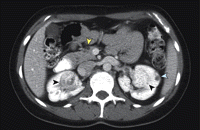
Figure 2. Scanner abdominal
Le scanner axial post contraste a révélé des carcinomes rénaux bilatéraux (flèche noire), des kystes rénaux ( flèche bleue) et des kystes pancréatiques (flèche jaune).
|
|
|
Des néphrectomies partielles bilatérales ont été exécutées pour réséquer de multiples RCCs le mois avant et après la résection de l’ELST. Dix-huit mois après la résection de l’ELST (19 mois après évaluation initiale au NIH), la patiente est revenus avec de nouvelles plaintes de maux de tête, de hoquets fréquents, et des difficultés pour avaler. L’IRM cranio-spinal a révélé à ce moment là le développement d'un kyste péritumoral associé à un hémangioblastome de l'obex (Figure 1). En vue d’alléger ses symptômes, la patiente a subi une résection de l'hémangioblastome de l'obex .L’IRM postopératoire a révélé que la résection de la tumeur et la résolution du kyste étaient complètes. Les signes et symptômes du tronc cérébral que présentaient la patiente ont été résolus (temps de suivi, 21 mois). Les scanners (CT) abdominaux répétés ont révélé des kystes rénaux stables, un RCCs, et des kystes pancréatiques. Les détails concernant les aspects cliniques, le diagnostic et le traitement lié à l’ELST de cette patiente ont été publiés précédemment.1,2
DISCUSSION
Maladie de von Hippel-Lindau
La maladie de von Hippel-Lindau est un syndrome de néoplasie autosomale dominante (prédominance de 1 sur 39 000 naissances vivantes) 3 résultant d'une mutation de la lignée germinale du gène VHL.4,5 Malgré son expression variable, la maladie de VHL possède une pénétration supérieure à 90% vers l’âge de 65 ans.6 Les patients atteints de la maladie de VHL sont prédisposés à développer des lésions spécifiques du système nerveux central et des lésions viscérales (Tableau 1).7,8 Dans le cas du système nerveux central, des tumeurs peuvent se développer, dont des hémangioblastomes et des ELST. Dans les viscères, des kystes rénaux, des RCC, des kystes pancréatiques, des tumeurs pancréatiques neuroendocriniennes, des phéochromocytomes, et des cystadénomes des organes reproducteurs annexes peuvent se développer. Avant la survenue d’une surveillance de routine et des recommandations de traitement mieux définies pour les lésions associées à la maladie de VHL (Tableau 2), la survie moyenne était de 50 ans 8,10 les causes fondamentales de décès étaient des complications liés aux hémangioblastomes du système nerveux central ou aux RCC.
|
|
|
|
Tableau 1. Diffusion approximative, âge de début, et la fréquence de lésions a associé à la maladie de von Hippel-Lindau (Adapté de Lonser et al7)
|
|
|
|
|
|
|
Tableau 2. Modalités de surveillance suggérées et fréquence des manifestations neurologiques de la maladie de von Hippel-Lindau (Adapté De Choyke et col. 9)
|
|
|
Diagnostic
Le diagnostic de VHL peut être établi par critères cliniques ou essai génétique. Les patients ayant une histoire famille de maladie de VHL et présentant un hémangioblastome du système nerveux central, carcimome rénal, phéochromocytome, ou tumeurs du sac de la poche endolymphatique correspondent aux critères du diagnostic de maladie de VHL. Environ 20% des patients ne possèdent pas d’histoire familiale mais remplissent les critères cliniques diagnostiques de maladie de VHL s’ils ont 2 ou plus hémangioblastomes du système nerveux central ou 1’hémangioblastome du système nerveux central et une tumeur viscérale associée à la maladie de VHL.10,11 fréquemment, les patients à risque subissent un test de mutation VHL de la lignée germinale germline.12
Le taux de détection de mutations VHL chez les patients ayant une histoire familiale de maladie de VHL avoisine les 100%. 13 Cependant, des mutations de novo chez les patients sans histoire familliale peuvent entraîner une mosaïque de la maladie dans laquelle quelques-uns mais pas tous les tissus portent la mutation .14 Ces patients peuvent être testés négatifs si leurs leucocytes du sang périphérique ne sont pas porteurs de la mutation gène VHL.
Pathogénie moléculaire
Le gène suppresseur de la tumeur de VHL était représenté sur une carte sur le bras court, du chromosome 3 par Seizinger et ses collègues 15 en 1988 et isolé par Latif et ses collègues 4 en 1993. Pareillement à notre patiente, la plupart des patients atteints de VHL héritent d’un gène VHL (allèle) avec une mutation de la lignée germinale venant du parent affecté et un gène VHL normal (phénotype sauvage) provenant du parent non affecté. Bien que toutes les cellules possèdent une mutation de la lignée germinale VHL , chez les patients héritant du trait, les tumeurs ne se forment que dans les cellules ayant perdu la fonction de l'allèle 16 de phénotype sauvage et se trouvant localisées dans des organes cibles spécifiques susceptibles de la maladie de VHL.7
Le gène VHL est largement exprimé dans les tissus, y compris ceux non affectés par la maladie de VHL.4 L’ARN messager de VHL encode la protéine VHL (pVHL). Au stade post translation, les complexes pVHL avec elongin B, elongin C, Rbx1, et cullin 2, formant une ligase de l'ubiquitin qui protéolyse la sous-unité du facteur inducteur d’hypoxie (HIF).7,17-20 Dans des circonstances normales, le HIF coordonne la réponse cellulaire à l’hypoxie à travers la régulation de la transcription. Le facteur inducteur d’hypoxie augmente le métabolisme cellulaire et accroît l'expression des facteurs angiogéniques et mitogéniques. Certains de ces facteurs comprennent un facteur de croissance endothéliale vasculaire VEGF),une chaîne de facteurs de croissance dérivés des plaquettes (PDGF), l’érythropoiétine, et transforment le facteur de croissance (TGF).7,18
Si la fonction pVHL est absente ou anormale, HIF peut stimuler constitutivement l'angiogénèse au moyen d’une augmentation des taux de VEGF ou PDGF 8,7,19 expliquant la nature vasculaire des tumeurs associées à la maladie de VHL.21 La perméabilité vasculaire de la tumeur accrue favorisée par VEGF peut constituer la cause de la formation fréquente d’oedèmes péritumoraux et de kystes dans la maladie de VHL. Le développement favorisé par HIF de boucles autocrines par la surproduction de TGF-_ ou d’érythropoiétine lié à la surexpression de leurs récepteurs réceptifs peuvent être sous-jacents à latumorogénèse.7,22,44
La tumorogénèse, indépendante de la régulation HIF, peut être provoquée ou augmentée uniquement si pVHL est absent ou anormal.7 Les cellules manquant de pVHL perdent la capacité de sortir du cycle cellulaire, ce qui peut constituer un événement précoce dans la tumorogénèse de la maladie de VHL.23 L’absence de pVHL peut continuer d’augmenter l’expression VEGF à travers la libération de la régulation de la transcription et de la post translation.24 Les cellules manquant de pVHL ne peuvent pas correctement assembler la matrice extracellulaire de fibronectine.25
Lésions associées à la maladie de VHL (à l'exclusion des Hémangioblastomes craniospinaux et des ELSTs)
RCC et kystes rénaux. Le carcinome rénal est le plus commun des néoplasmes malins dans la maladie de VHL (Tableau 1). Un RCC ou un kyste peut être trouvé chez 60% des patients. Les kystes rénaux sont typiquement asymptomatiques et nécessitent rarement un traitement. Cependant, les kystes complexes ont besoin de surveillance en série parce qu'ils hébergent souvent des composants solides de RCC. Bien que les petits RCC aient tendance à être de bas grade et peu invasifs, le taux de croissance de ces lésions est très variable.26 Parce que les RCC peuvent rester asymptomatiques pendant de longues périodes, l’imagerie en série est utile pour un diagnostic précoce (Tableau 2). Rarement, dans les cas les plus avancés, les patients présentent une hématurie, une douleur du flanc ou une masse dans le flanc. Quelques cliniciens recommandent la résection des RCC épargnant les néphrons quand la plus grande tumeur atteint un diamètre maximal de 3 centimètres. La chirurgie épargnant les néphrons ou les reins est fondée sur la capacité de préserver la fonction rénale en réduisant le risque de métastase. Walther et col.26 ont rapporté que la chirurgie ménageant les reins pour les RCC mesurant moins de 3 centimètres n'était pas associée à des métastases ou à un besoin de transplantation rénale ou de dialyse. L'énucléation tumorale ou la néphrectomie partielle peuvent être nécessaires à la résection des tumeurs supérieures à 3 centimètres. Nous explorons des traitements percutanés moins invasifs de RCC.
Tumeurs neuroendocriniennes et kystes pancréatiques. 35% à 70% des patients atteints de la maladie de VHL développent une tumeur neuroendocrinienne ou un kyste pancréatique (Tableau 1). Les kystes pancréatiques sont généralement asymptomatiques et n'exigent pas de traitement. Les tumeurs neuroendocriniennes pancréatiques sont souvent non fonctionnelles et asymptomatiques mais peuvent devenir malignes dans jusqu'à 8% des cas.27
Ces tumeurs sont identifiées par scanner abdominal post contraste ou IRM (Tableau 2). Les données récentes indiquent que le timing optimal de la résection chirurgicale des tumeurs pancréatiques neuroendocriniennes peut être fondé sur la taille de la tumeur, le statut de la mutation exon 3 VHL, et le temps de doublement de la tumeur.27
Phéochromocytome. Les Phéochromocytomes peuvent être multiples et bilatéraux naturellement dans la maladie de VHL (Tableau 1). On peut également les trouver en tant que paragangliomes extra-surrénaux dans le corps carotidien, le glomus jugulaire , et les tissus périaortiques. Cinq pour cent des phéochromocytomes sont malins.28 Les fréquents résultats cliniques associés aux phéochromocytomes incluent une hypertension intermittente ou soutenue, des palpitations, de la tachycardie, des maux de tête, de la sueur épisodique, de la pâleur, et des nausées, mais 30% des patients atteints de phéochromocytome sont asymptomatiques.28 Le diagnostic de phéochromocytome est établi par des études de laboratoire et d’imagerie (Tableau 2). L’évaluation préopératoire est critique chez les patients atteints de la maladie de VHL afin de prévenir une crise d’hypertension péri-opératoire. Une intervention précoce avec chirurgie d’épargne cortico-surrénale entraîne une faible récurrence et une indépendance à long terme aux corticostéroïdes.
Cystadénomes des organes reproducteurs annexes. Les cystadénomes de l'épididyme et du ligament large sont des lésions bénignes pouvant fréquemment être trouvées chez les patients atteints de la maladie de VHL (Tableau 1). Ils sont souvent bilatéraux et multiples. Ils peuvent être identifiés par ultrason (épididyme) ou scanner abdominal (ligament large). Parce que ces lésions sont bénignes et plus souvent asymptomatiques, on les gère de façon à les conserver.
Hémangioblastomes rétiniens. Les hémangioblastomes rétiniens sont souvent bilatéraux et multifocaux dans la maladie de VHL (Tableau 1). Ils peuvent survenir dans la périphérie, et soit à proximité soit directement sur le disque optique. En dépit de leur caractère fréquemment asymptomatique, ils peuvent causer des symptômes visuels par croissance progressive, par oedème ou par développement d’exsudat solide. L’ophtalmoscopie avec dilatation de l'iris permet l’identification de la plupart des hémangioblastomes rétiniens. Le diagnostic et traitement précoces (photocoagulation et cryothérapie) de tumeurs périphériques peuvent prévenir la perte visuelle.
Hémangioblastomes craniospinaux.
Épidémiologie. L’hémangioblastome craniospinal est la tumeur la plus commune associée à la maladie de VHL (Tableau 1). Soixante à 80% des patients atteints de la maladie de VHL développent un hémangioblastome du système nerveux central au niveau du cervelet, du tronc cérébral ou de la moelle épinière.7 La quasi totalité de ces patients (90%) développe des hémangioblastomes multiples. Malgré leur histologie bénigne, les hémangioblastomes peuvent causer une morbi-mortalité considérable dans la maladie de VHL. Les hémangioblastomes du système nerveux central (à l'extérieur de la rétine) sont trouvés quasi exclusivement (95% des tumeurs) dans le tronc cérébral, le cervelet ou la moelle épinière.7,29,30
Résultats de l’imagerie. L’IRM avec augmentation de contraste est la méthode d’imagerie la plus sensible et la plus précise pour détecter et surveiller les hémangioblastomes. Les hémangioblastomes augmentent vivement et discrètement les séquences d’ IRM pondérée T1. Même de petites tumeurs (2 mm de diamètre) sont détectées de façons fiables et caractérisées précisément sur des études en série. L’utilisation de l’IRM est critique pour détecter le développement ou les changements dans l’oedème péri tumoral et les kystes contenant des fluides ont atténué la récupération de l'inversion et les séquences pondérées T2.
Présentation des signes et symptômes. Les signes et symptômes liés aux hémangioblastomes du système nerveux central sont attribuables à la région anatomique où ils apparaissent. Les hémangioblastomes cérébelleux provoquent mal de tête (75% des patients), ataxie de la démarche (55%), dysmétrie (29%), hydrocéphalie (28%), et nausée ou vomissements (28%).31 Les hémangioblastomes du tronc cérébral provoquent hypoesthésie (55%), ataxie de la démarche (22%), dysphagie (22%), hyperréflexie (22%) et mal de tête (11%).7,32 Les hémangioblastomes de la moelle épinière provoquent hypoesthésie (83%), faiblesse (65%), ataxie de la démarche (65%), hyperréflexie (52%) et douleur (17%).7
Histoire naturelle. Afin de mieux définir l’histoire naturelle des hémangioblastomes associés à la maladie deVHL, Wanebo et col. 30 ont examiné l’imagerie et les caractères cliniques de 160 patients consécutifs atteints de la maladie de VHL qui hébergent 655 hémangioblastomes du système nerveux central suivi au NIH (moyenne [SD] temps de suivi, 21 [27] mois). La circonstance clinique était dynamique et on a trouvé que les tumeurs possédaient des modèles de croissance variable. Bien que la formation du symptôme semblait associée à la taille de la tumeur, au taux de croissance de la tumeur et à la présence de kyste péri-tumoral, aucun seuil fiable concernant la taille ou la croissance de la tumeur ne pourrait être identifié comme annonçant l’apparition de symptômes et la nécessité d’un traitement.
Ammerman et col 33 ont étudié le modèle de croissance des hémangioblastomes du système nerveux central chez 19 patients atteints de la maladie de VHL ayant effectué des imageries en série pendant au moins 10 années. Les hémangioblastomes ont grandi dans un modèle de croissance saltatoire qui consiste en périodes de croissance rapide suivies par des périodesde repos (97% de 143 tumeurs). Les hémangioblastomes avaient en moyenne 1,85 périodes de repos entre des périodes de croissance avant de devenir symptomatiques et de nécessiter une résection. Les périodes de croissance ont duré en moyenne (SD) 13 (15) mois, tandis que les périodes de repos étaient en moyenne de 25 (19) mois. Presque tous les émangioblastomes (97%) ont montré une progression radiographique, mais seulement 50% ont nécessité un traitement pour la survenue de symptôme. Presque la moitié des tumeurs (45%) nécessitant une résection (symptomatique) n'apparaissait pas sur l’IRM initial.
Formation de kyste péri-tumoral. Bien que le développement des signes et des symptômes puissent être parfois directement attribués à l'effet de masse de l’hémangioblastome, le dysfonctionnement neurologique est les plus souvent causé par l'effet de masse combiné de la tumeur et du kyste péri-tumoral associé (Figure 1). La plupart des hémangioblastomes symptomatiques cérébelleux et du tronc cérébral (70%) est associée à un kyste péri-tumoral, et plus de 90% des hémangioblastomes symptomatiques spinaux sont associé à un kyste péri-tumoral (syringomyélie).32,33 Dans de tels cas, le kyste explique le volume de la masse pesante. Précisément, le volume moyen du kyste cérébelleux était 4 fois plus grand que celui de la tumeur associée, et celui du tronc cérébral était 12 fois plus grand que celui de la tumeur associée chez des patients symptomatiques. Inversement, une petite fraction des hémangioblastomes asymptomatiques du cervelet, du tronc cérébral ou de la moelle épinière (5%-10%) est associée à des kystes péri-tumoraux.
La compréhension du mécanisme sous-jacent au développement et à la propagation du kyste péri-tumoral avec les hémangioblastomes associés à la maladie de VHL a conduit à plusieurs perspectives cliniques critiques (Figure 3).34,35 Puisque la tumeur est la source de la formation d'oedème/kyste péritumoral, la résolution d'oedème/kyste après ablation de la tumeur se produit de façon fiable et le traitement n'exige pas la résection du mur du kyste ou fenêtrage. La réduction de la perméabilité vasculaire de la tumeur pourrait être salutaire, et les progrès cliniques utilisant les traitements anti-VEGF ont été associés à une réduction de l'oedème bien que n’ayant aucun effet sur la taille de la tumeur.36,37 L’accroissement de la perméabilité vasculaire peut être nuisible, et des études ont montré que les radiations induisaient des augmentations transitoires de la perméabilité vasculaire pouvant entraîner la formation d’un oedème et d’un kyste péri-tumoral, suggérant une irradiation judicieuse des hémangioblastomes avec d’importants oedème/kystes péri-tumoraux.38 Finalement, les kystes péri-tumoraux peuvent stopper leur croissance quand la surface du mur du kyste est suffisamment grande pour absorber le fluide en excès et se mettre au repos et rester asymptomatique. La preuve par imagerie de la formation de l’oedème et du kyste chez les patients asymptomatiques atteints de la maladie de VHL ne sont pas une indication absolue pour la chirurgie.
|
|
|
Figure 3. Progression de l’hémangioblastome associé à la formation péri-tumorale d’œdème et de kyste
|
|
|
Caractéristiques de la tumeur. Grossièrement, les hémangioblastomes apparaissent avec une coloration rouge clair ou orange/jaune et sont invariablement associés à une vascularisation intense (Figure 3). Histologiquement, les hémangioblastomes possèdent des traits caractéristiques comprenant la prolifération des cellules du stroma et endothéliales (Organisation Mondiale de la Santé grade 1) (Figure 3).39 Les cellules endothéliales forment des canaux vasculaires autour des cellules néoplasiques du stroma.40 Les cellules du stroma possèdent de nombreuses vacuoles contenant des lipides conduisant à la morphologie cellulaire claire semblable au RCC. Puisque les patients atteints de la maladie de VHL présentent souvent des RCC contemporains pouvant se métastaser dans les tissus ou les hémangioblastomes du système nerveux central, 39,41 la différenciation immunohistochimique entre RCC et hémangioblastome peut être nécessaire.
Origine du développement. En se fondant sur les résultats embryologiques et la preuve de l’hématopoïèse intratumorale et la formation de la cellule endothéliale, nous avons supposé que les hémangioblastomes peuvent provenir d'une cellule embryologique capable de former du sang et des vaisseaux. Cette théorie n'était pas directement testable jusqu'en 1998, lorsque Choi et col. 42 ont caractérisé un précurseur commun de cellule embryologique qui était provisoirement présent durant le développement du mésoderme et était capable de former du sang et des cellules endothéliales. Ils ont défini cette cellule embryologique multipotente comme hémangioblaste. Les investigations ultérieures ont uniquement identifié l'hémangioblaste embryonnaire par coexpression de brachyurie, Flk1 (récepteur VEGF 2), et leucémie de la cellule souche (Scl).43 Comme l’hémangioblaste embryonnaire, les cellules néoplasiques du stroma dans les hémangioblastomes ont coexprimé une brachyurie , Flk1, et Scl.44
Traitement. La Résection complète des hémangioblastomes est curative et la plupart des hémangioblastomes craniospinaux peuvent être réséqués sans risque.31,32,45 En raison de la multiplicité des hémangioblastomes du système nerveux central, et leur taux de croissance étant imprévisible, la chirurgie est généralement réservée jusqu'à ce que les symptômes associés surviennent.30,33 En utilisant cette stratégie, la plupart des malades atteints de la maladie de VHL peut maintenir une excellente fonction neurologique et la résection chirurgicale inutile peut être évitée.33
La radiochirurgie stéréotaxique a été étudiée en tant qu’option thérapeutique potentielle. 38,46 Les petits hémangioblastomes non associés aux kystes péri-tumoraux peuvent répondre au mieux à la radiothérapie. Bien que certaines études aient établi le succès de l'usage de la radiochirurgie fondée sur la stabilité de la taille de la tumeur, le manque de progression de l’hémangioblastome dans ces cas peut représenter une période de repos et non une réponse au traitement. 31,33 Une estimation à plus long terme sur davantage de patients est nécessaire pour déterminer l'efficacité de ce traitement et le risque potentiel de développer de nouvelles néoplasies chez les patients présentant une haploinsuffisance constitutionnelle.
Tumeurs du sac endolymphatique
Épidémiologie. Les tumeurs du sac endolymphatique ont d’abord été établies comme une entité pathologique distincte par Heffner 47 en 1989 et ont été reconnues comme élément du syndrome de la maladie de VHL par Manski et col 48 en 1997. On peut trouver la preuve par imagerie d’une ELST chez environ 10% à 15% des patients atteints de la maladie de VHL, et 30% des patients atteints de VHL avec une ELST développent des ELST bilatérales.48 En dépit de leur histologie bénigne, les ELST sont des tumeurs localement invasives causant une morbidité audiovestibulaire, y compris perte auditive, vertige, tintement d'oreilles, et douleur auditive.1,48,49
Résultats de l’imagerie. L’identification optimale des ELST exige scanner et IRM de haute résolution. Sur les images du scanner, les ELST sont des tumeurs à faible densité tissulaire étendant, détruisant et incorporant l'os temporal adjacent. De petites érosions osseuses adjacentes à l'aqueduc vestibulaire peuvent être visibles sur le scanner comme résultat radiographique initial d'une ELST. Sur les IRM, les ELST peuvent avoir une considérable hétérogénéité parce que les kystes et hémorragies internes, l’os temporal incorporé, les granulomes de cholestérol, et les vides du courant vasculaire peuvent être présents à des degrés variables. L’hémorragie intralabyrinthique, un caractère commun de l’imagerie associé aux ELST, apparaît comme une hyperintensité pondérée T1 dans le vestibule, la cochlée, ou les canaux semi-circulaires, distincte du site de la tumeur.
Signes et symptômes présents. Les patients atteints de maladie deVHL avec preuve par imagerie d'une ELST présentent perte auditive (95% des patients), tintement d'oreilles (90%), vertige ou déséquilibre (66%), plénitude auditive (30%), et parésie faciale (8%).48 La perte auditive peut se produire soit de façon aiguë (86%) soit progressivement sur plusieurs années (14%).48 Généralement, la perte auditive est irréversible une fois survenue, et typiquement cela se produit en début de vie (Tableau 1).48 Comme dans notre cas, les patients rapportent habituellement que cette perte auditive coïncide avec l’exacerbation de symptômes vestibulaires.2 Un nombre considérable de patients atteints de la maladie de VHL avec symptômes vestibulocochléaires (59%) ne possèdent aucune preuve par imagerie des ELST.48,49 L'étiologie de ces manifestations cliniques peut être due à un ELST microscopique ou à une hyperplasie de l' épithélium endolymphatique chez les patients atteints de maladie de VHL.1,50
Mécanismes de perte auditive. Afin de définir les mécanismes associés à ELST, sous-jacents à la physiopathologie audiovestibulaire, une étude prospective menée sur des patients atteints de la maladie de VHL avec des ELST a été effectuée.51 Les données cliniques et audiologiques ont été corrélées avec scanner en série et IRM afin d’élucider les mécanismes sous-jacents du dysfonctionnement audiovestibulaire. Bien que l'invasion de la tumeur de la capsule otique (revêtement osseux de l'appareil de l'oreille interne) (18% des patients) ait été associée à de grands ELST et ait causé une perte auditive (100% des patients avec invasion de la capsule otique), la majorité des patients (82%) présentaient de petits ELST n’ayant pas envahi la capsule otique. Dans ce groupe de patients, la perte auditive était encore présente (91% des oreilles affectées) et soit s’est développée soudainement (43%) soit progressivement (48%). La perte auditive soudaine chez ces patients a correspondu à la preuve par IRM d'hémorragie intralabyrinthique, mais nous n’avons pas observé d'hémorragie chez les patients à audition graduelle ou normale. La taille de la tumeur n'a pas été associée au développement des symptômes audiovestibulaires.
Les résultats de cette étude soutiennent 3 mécanismes distincts de morbidité audiovestibulaire associés à ELST, dont invasion directe de la capsule otique par la tumeur, hémorragie intralabyrinthique, et hydropsie endolymphatique. Premièrement, l’érosion directe d’ ELST dans l'oreille interne peut entraîner la destruction du labyrinthe membraneux interrompant le flux endolymphatique, causant perte de l'audition et vestibulopathie. Deuxièmement, l’hémorragie intralabyrinthique aiguë, telle qu’observée chez les patients avec perte auditive soudaine sans invasion de la capsule otique, indique que l'hémorragie spontanée associée à ELST guidée dans le labyrinthe par le conduit endolymphatique peut sous-tendre la soudaine perte auditive dans ces cas. Ce mécanisme est entretenu par la présence d’hémosidérine dans le labyrinthe membraneux chez un patient sourd atteint de la maladie de VHL avec une ELST à l’autopsie.1 Finalement, chez les patients présentant une perte auditive graduelle, aucune hémorragie n'a été identifiée et le complexe symptomatique était semblable à celui de notre patient et a imité celui de la maladie de Ménière. Le développement de l'hydropsie endolymphatique est une autre conséquence possible d'une petite ELST. L’hydropsie représente une augmentation du volume de l'endolymphe, la régulation que l’on pense être la fonction primaire du sac endolymphatique. L'endolymphe étant produite à l'origine dans la cochlée, l’hydropsie chez les patients présentant des ELST pourraient se développer au moyen de la résorption de l'endolymphe affaiblie ou en raison de la production excessive de fluide dans le labyrinthe membraneux. La production excessive de fluide par l'ELST pourrait être analogue à la formation d’oedème péri-tumoral ou de kystes associés avec des hémangioblastomes du système nerveux central et des tumeurs viscérales dans la maladie de VHL.1,7
Caractéristiques de la tumeur. Grossièrement, les ELST peuvent apparaître en couleur rouge clair ou sombre. Histologiquement, les ELST sont des néoplasies glandulaires papillaires cystiques bien vascularisées, bordées par une rangée de cellules cuboïdales. Le pléomorphisme proéminent n'est pas observé et l'activité mitotique est absente. 47 Les caractères ultrastructurels de ces tumeurs sont composés de l’épithélium du sac endolymphatique.47 Malgré le manque de caractères malins associés aux ELST, ils ont été répertoriés en tant qu’adénocarcinomes de bas grade (plutôt qu'adénomes) d'origine du sac endolymphatique.47 Une preuve récente indique que les ELST proviennent d’une partie de l'aqueduc vestibulaire du sac endolymphatique et du système du conduit.52
Origine du développement. Des études ont révélé des proliférations multifocales de cellules épithéliales sans VHL à travers le conduit et le sac endolymphatiques chez les patients atteints de la maladie de VHL. Ces proliférations peuvent représenter des structures de potentiels précurseurs du développement d’une tumeur franche.50 L’abondance de structures de précurseurs détectée dans le sac et le conduit endolymphatiques chez un patient atteint de la maladie de VHL sans ELST à l’autopsie suggère que la plupart des structures de précurseur n’évoluent pas en tumeurs pendant la vie d'un patient.53
Traitement. Comme la morbidité audiovestibulaire est fréquemment associée à des tumeurs très petites et puisqu’une perte auditive irréversible peut survenir soudainement indépendamment de la dimension de la tumeur, l'intervention chirurgicale précoce peut être justifiée. La résection complète des ELST est curative, peut alléger la symptomatologie vestibulaire, et peut être effectuée avec conservation de l’audition et morbidité minime.2,54,55 Il est justifié d’intervenir tôt chez les patients atteints de la maladie de VHL, d’établir un diagnostic promptement, fondé sur des résultats cliniques et soutenu par scanner et IRM à haute résolution en vue de détecter les petites ELST et les hémorragies intra-labyrinthiques. La découverte d'une ELST peut, après avoir évalué les risques potentiels, induire la chirurgie en vue d’améliorer les symptômes et prévenir la progression de perte auditive.
CONCLUSIONS
Les investigations récentes sur les lésions du système nerveux central associées à la maladie de VHL ont fourni une perspective concernant l'origine et le développement de ces tumeurs. Les données venant de l'imagerie et des protocoles de surveillance clinique ont fourni des perspectives concernant l’histoire naturelle des ELST et des hémangioblastomes associés à la maladie de VHL. En raison de leurs histoires naturelles différentes, les stratégies de gestion optimale pour les 2 manifestations neurologiques de la maladie de VHL sont différentes.
Informations sur les auteurs
Correspondance: Russell R. Lonser, MD, Surgical Neurology Branch, National Institute of Neurological Disorders and Stroke, 10 Center Dr, Bldg 10, Room 5D37, National Institutes of Health, Bethesda, MD 20892-1414 (lonserr{at}ninds.nih.gov).
Contributions d’auteur : le Dr Lonser a eu accès à toutes les données de l’étude et prend la responsabilité de l’intégrité des données et de la précision de l’analyse des données.
Concept d’étude et conception : Butman, Lonser
Acquisition des données : Butman, Linehan, Lonser
Analyse et interprétation des données : Butman, Linehan, Lonser.
Rédaction du manuscrit : Butman, Lonser.
Révision critique du manuscrit pour le contenu intellectuel important : Butman, Linehan, Lonser.
Financement obtenu : Butman, Linehan, Lonser.
Soutien administratif, technique, ou matériel : Butman, Linehan, Lonser.
Supervision de l’étude : Butman, Lonser.
Révélations financières : Aucune.
Financement/soutien : Cette recherche a été soutenue par l’Intramural Research Program of the Clinical Center, le National Institute of Neurologic Disorders Stroke, et le National Cancer Institute at the National Institutes of Health.
Role du Sponsor : Les organisations de financement n’ont joué Aucun rôle dans la conduite et la conception de l’étude ; dans la collection, analyse, et interprétation des données, ou la préparation, revue ou approbation du manuscrit.
Affiliations des auteurs: Département de radiologie diagnostique, Clinical Center of the National Institutes of Health, Bethesda, Maryland (Dr Butman);Urologic Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health (Dr Linehan); and Surgical Neurology Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health (Dr Lonser).
FMC disponible en ligne sur www.jamaarchivescme.com et quiz p 1364.
Editeur de la Section des séances Scientifiques : David S. Cooper, MD, Editeur contribuant.
BIBLIOGRAPHIE
| |
1. Lonser RR, Kim HJ, Butman JA, Vortmeyer AO, Choo DI, Oldfield EH. Tumors of the endolymphatic sac in von Hippel-Lindau disease. N Engl J Med. 2004; 350(24):2481-2486.
PUBMED
2. Kim HJ, Butman JA, Brewer C, et al.. Tumors of the endolymphatic sac in patients with von Hippel-Lindau disease: implications for their natural history, diagnosis, and treatment. J Neurosurg. 2005;102 (3):503-512.
PUBMED
3. Neumann HP, Wiestler OD. Clustering of features of von Hippel-Lindau syndrome: evidence for a complex genetic locus. Lancet. 1991;337(8749): 1052-1054.
PUBMED
4. Latif F, Tory K, Gnarra J, et al.. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260(5112):1317-1320.
FREE FULL TEXT
5. Wait SD, Vortmeyer AO, Lonser RR, et al.. Somatic mutations in VHL germline deletion kindred correlate with mild phenotype. Ann Neurol. 2004; 55(2):236-240.
PUBMED
6. Maher ER, Iselius L, Yates JR, et al.. Von Hippel-Lindau disease: a genetic study. J Med enet. 1991;28(7):443-447.7. Lonser RR, Glenn GM, Walther M, et al.. von Hippel-Lindau disease. Lancet. 2003;361(9374):2059-2067.
PUBMED
8. Richard S, Campello C, Taillandier L, Parker F, Resche F.. Haemangioblastoma of the central nervous system in von Hippel-Lindau disease: French VHL Study Group. J Intern Med. 1998;243(6):547-553.
PUBMED
9. Choyke PL, Glenn GM, Walther MM, Patronas NJ, Linehan WM, von Zbar B. Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology. 1995;194(3):629-642.
FREE FULL TEXT
10. Lamiell JM, Salazar FG, Hsia YE. von Hippel-Lindau disease affecting 43 members of a single kindred. Medicine (Baltimore). 1989;68(1):1-29.
PUBMED
11. Melmon KL, Rosen SW. Lindau’s disease. Am J Med. 1964;36:595-617.
PUBMED
12. Woodward ER, Wall K, Forsyth J, Macdonald F, Maher ER. VHL mutation analysis in patients with isolated central nervous system haemangioblastoma.Brain. 2007;130(pt 3):836-842.
FREE FULL TEXT
13. Stolle C, Glenn G, Zbar B, et al.. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998; 12(6):417-423.
PUBMED
14. Sgambati MT, Stolle C, Choyke PL, et al.. Mosaicism in von Hippel-Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am J Hum Genet. 2000;66 (1):84-91.
PUBMED
15. Seizinger BR, Rouleau GA, Ozelius LJ, et al.. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988;332(6161):268-269.
PUBMED
16. Knudson AGJ, Strong LC. Mutation and cancer: neuroblastoma and pheochromocytoma. Am J Hum Genet. 1972;24(5):514-532.
PUBMED
17. Duan DR, Pause A, Burgess WH, et al.. Inhibition of transcription elongation by the VHL tumor suppressor protein [see comments]. Science. 1995; 269(5229):1402-106.
FREE FULL TEXT
18. Kaelin WG Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2 (9):673-682.
PUBMED
19. Maxwell PH, Wiesener MS, Chang GW, et al.. The tumour suppressor protein VHL targets hypoxiainducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271-275.
PUBMED
20. Pause A, Lee S, Worrell RA, et al.. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, amember of the Cdc53 family of proteins. Proc Natl Acad Sci U S A. 1997; 94(6):2156-2161.
FREE FULL TEXT
21. Carmeliet P, Dor Y, Herbert JM, et al.. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394 (6692):485-490.
PUBMED
22. Reifenberger G, Reifenberger J, Bilzer T, Wechsler W, Collins VP. Coexpression of transforming growth factor-alpha and epidermal growth factor receptor in capillary hemangioblastomas of the centralc nervous system. Am J Pathol. 1995;147(2):245-250.
PUBMED
23. Pause A, Lee S, Lonergan KM, Klausner RD. The von Hippel-Lindau tumor suppressor gene is required for cell cycle exit upon serum withdrawal. ProcNatl Acad Sci U S A. 1998;95(3):993-998.
FREE FULL TEXT
24. Gnarra JR, Zhou S, Merrill MJ, et al.. Posttranscriptional regulation of vascular endothelialgrowth factor mRNA by the product of the VHL tumor suppressor gene. Proc Natl Acad SciUS A. 1996; 93(20):10589-10594.
25. Ohh M, Yauch RL, Lonergan KM, et al.. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1(7):959-968.
PUBMED
26. Walther MM, Choyke PL, Glenn G, et al.. Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol. 1999;161 (5):1475-1479.
PUBMED
27. Blansfield JA, Choyke L, Morita SY, et al.. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery. 2007;142(6):814-818.
PUBMED
28. Walther MM, Reiter R, Keiser HR, et al.. Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol. 1999;162(3 pt 1): 659-664.
PUBMED
29. Browne TR, Adams RD, Roberson GH. Hemangioblastoma of the spinal cord: review and report of five cases. Arch Neurol. 1976;33(6):435-441.
FREE FULL TEXT
30. Wanebo JE, Lonser RR, Glenn GM, Oldfield EH. The natural history of hemangioblastomas of the central nervous system in patients with vonc Hippel-Lindau disease. J Neurosurg. 2003;98(1):82-94.
PUBMED
31. Jagannathan J, Lonser RR, Smith R, DeVroom HL, Oldfield EH. Surgical management of cerebellar hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2008;108(2):210-222.
PUBMED
32. Lonser RR, Weil RJ, Wanebo JE, DeVroom HL, Oldfield EH. Surgical management of spinal cord hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98(1):106-116.
PUBMED
33. Ammerman JM, Lonser RR, Dambrosia J, Butman JA, Oldfield EH. Long-term natural history of hemangioblastomas in patients with von Hippel-Lindau disease: implications for treatment. J Neurosurg. 2006; 105(2):248-255.
PUBMED
34. Lonser RR, Vortmeyer AO, Butman JA, et al.. Edema is a precursor to central nervous system peritumoral cyst formation. Ann Neurol. 2005;58(3):392-399.
PUBMED
35. Lohle PN, van Mameren H, Zwinderman KH, Teepen HL, Go KG, Wilmink JT. On the pathogenesis of brain tumour cysts: a volumetric study of tumour, oedema and cyst. Neuroradiology. 2000; 42(9):639-642.
PUBMED
36. Aiello LP, George DJ, Cahill MT, et al.. Rapid and durable recovery of visual function in a patient with von Hippel-Lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor su5416. Ophthalmology. 2002;109(9):1745-1751.
PUBMED
37. Girmens JF, Erginay A, Massin P, Scigalla P, Gaudric A, Richard S. Treatment of von Hippel-Lindau retinal hemangioblastoma by the vascular endothelial growth factor receptor inhibitor SU5416 is more effective for associated macular edema than for hemangioblastomas. Am J Ophthalmol. 2003; 136(1):194-196.
PUBMED
38. Niemela M, Lim YJ, Soderman M, Jaaskelainen J, Lindquist C. Gamma knife radiosurgery in 11 hemangioblastomas. J Neurosurg. 1996;85(4): 591-596.
PUBMED
39. Aldape KD, Plate KH, Vortmeyer AO, Zagzag D, Neumann HPH. Haemangioblastoma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2007:184-186.40. Vortmeyer AO, Gnarra JR, Emmert-Buck MR, et al.. von Hippel-Lindau gene deletion detected in the stromal cell component of a cerebellar hemangioblastoma associated with von Hippel-Lindau disease. Hum Pathol. 1997;28(5):540-543.
PUBMED
41. Jarrell ST, Vortmeyer AO, Linehan WM, Oldfield EH, Lonser RR. Metastases to hemangioblastomas in von Hippel-Lindau disease. J Neurosurg. 2006; 105(2):256-263.
PUBMED
42. Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998;125 (4):725-732.
ABSTRACT
43. Huber TL, Kouskoff V, Fehling HJ, Palis J, Keller G. Haemangioblast commitment is initiated in the primitive streak of the mouse embryo. Nature. 2004; 432(7017):625-630.
PUBMED
44. Park DM, Zhuang Z, Chen L, et al.. von Hippel-Lindau disease-associated hemangioblastomas are derived from embryologic multipotent cells. PLoS Med. 2007;4(2):e60.
PUBMED
45. Weil RJ, Lonser RR, DeVroom HL, Wanebo JE, Oldfield EH. Surgical management of brainstem hemangioblastomasin patients with von Hippel-Lindaudisease. J Neurosurg. 2003;98(1):95-105.
PUBMED
46. Patrice SJ, Sneed PK, Flickinger JC, et al.. Radiosurgery for hemangioblastoma: results of a multiinstitutional experience. Int J Radiat Oncol Biol Phys. 1996;35(3):493-499.
PUBMED
47. Heffner DK. Low-grade adenocarcinoma of probable endolymphatic sac origin: a clinicopathologic study of 20 cases. Cancer. 1989;64(11):2292-2302.
PUBMED
48. Manski TJ, Heffner DK, Glenn GM, et al.. Endolymphatic sac tumors: a source of morbid hearing loss in von Hippel-Lindau disease. JAMA. 1997;277 (18):1461-1466.
FREE FULL TEXT
49. Choo D, Shotland L, Mastroianni M, et al.. Endolymphatic sac tumors in von Hippel-Lindau disease. J Neurosurg. 2004;100(3):480-487.
PUBMED
50. Glasker S, Lonser RR, Tran MG, et al.. Effects of VHL deficiency on endolymphatic duct and sac. Cancer Res. 2005;65(23):10847-10853.
FREE FULL TEXT
51. Butman JA, Kim HJ, Baggenstos M, et al.. Mechanisms of morbid hearing loss associated with tumors of the endolymphatic sac in von Hippel-Lindau disease. JAMA. 2007;298(1):41-48.
FREE FULL TEXT
52. Lonser RR, Baggenstos M, Kim HJ, Butman JA, Vortmeyer AO. The vestibular aqueduct: site of origin of endolymphatic sac tumors. J Neurosurg. 2008; 108(4):751-756.
PUBMED
53. Glasker S, Li J, Xia JB, et al.. Hemangioblastomas share protein expression with embryonal hemangioblast progenitor cell. Cancer Res. 2006;66(8):4167-4172.
FREE FULL TEXT
54. Megerian CA, Haynes DS, Poe DS, Choo DI, Keriakas TJ, Glasscock ME III. Hearing preservation surgery for small endolymphatic sac tumors in patients with von Hippel-Lindau syndrome. Otol Neurotol. 2002;23(3):378-387.
PUBMED
55. Rodrigues S, Fagan P, Turner J.. Endolymphatic sac tumors: a review of the St. Vincent’s hospital experience. Otol Neurotol. 2004;25(4):599-603.
PUBMED
ARTICLE EN RAPPORT
Cette semaine dans le JAMA-Français
JAMA. 2008;300:1273.
Texte Complet
|