Innate Immunity in Alzheimer's Disease
A Model for Local Inflammatory Reactions
- Kinsmen Laboratory of Neurological Research, Department of Psychiatry University of British Columbia, Vancouver, BC V6T 1Z3
- Address correspondence to EGM. E-mail mcgeer{at}interchange.ubc.ca; fax 604-822-7086.
Abstract
Over the past fifteen years, evidence has been accumulating that there is a chronic inflammatory reaction in areas of the brain affected by Alzheimer's disease. Chronic inflammation, which arises in reaction to an underlying pathology, represents a threat in its own right, wherever it may occur, and can in fact surpass primary affronts upon tissues. The brain, however, is particularly vulnerable because neurons are generally irreplaceable. In the case of Alzheimer's disease, inflammatory processes thus have the potential for turning a relatively slowly progressing condition into one characterized by rapid neurodegeneration.
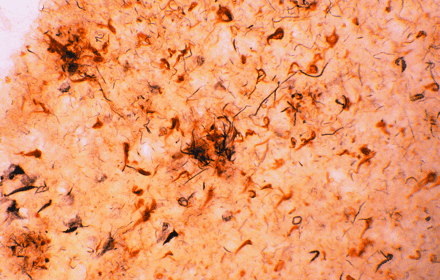
Amyloid plaques (large extracellular deposits) and neurofibrillary tangles (dense intracellular aggregates) are well-known
histological indicators of Alzheimer's disease. Intriguingly, these pathological hallmarks contain all of the major complement
proteins that are normally associated with immune activation. The afflicted brain tissue shown here has been stained with
antibodies specific for complement proteins.
Introduction
Evidence for the importance of inflammation in Alzheimer's disease comes from two directions. First, immunohistochemical and molecular biological studies reveal the sharp induction of inflammatory mediators in brain regions affected by Alzheimer's disease. In addition, approximately twenty epidemiological studies indicate that nonsteroidal anti-inflammatory drugs (NSAIDs) markedly reduce the age-related prevalence of Alzheimer's disease. Nevertheless, acceptance for the role of inflammation in Alzheimer's disease is still far from universal.
One basis for skepticism comes from an academic adherence to the classical definition of inflammation according to the four “cardinal signs” described by Celsus in the first century a.d., namely, calor, rubor, tumor, and dolor (i.e., heat, redness, swelling, and pain). These criteria, however, are deceptive in that they represent the consequences of a secondary physiological reaction in which inflammatory mediators expand circulation and cause vessels to leak serum into tissues. The brain is exempt from these classical criteria because such leakage is precluded by the blood brain barrier so that the brain does not react by swelling (tumor). Moreover, the brain lacks sensory fibers so that detection of dolor, calor, and rubor is also impossible. Neuroinflammation such as occurs in Alzheimer's disease is a silent process.
A second reason why many have been loath to accept the idea that a local immune reaction occurs in Alzheimer's disease is that the attention of immunologists has for decades been rather narrowly focused on the adaptive immune system in autoimmunity. In autoimmune disorders, lymphocytes are cloned against a self-epitope with the result that tissues can be very specifically attacked by antibodies and T-cell–mediated reactions. Indeed, in a wide variety of diseases (e.g., systemic lupus erythematosus), autoimmunity becomes a major pathological process. But besides the adaptive immune system and its occasional perversion into processes of disease, there is another immune system that we all possess, a fact that often goes underappreciated—if not totally ignored. Distinct from adaptive immunity, which is the invention of higher vertebrates, a phylogenetically ancient set of recognition mechanisms constitutes the innate immune system, which is in fact a vigorous first line of defense not only in humans, but also in invertebrates and even in plants.
The innate immune system is less sophisticated in its selectivity than the adaptive immune system, and may more readily confuse friend and foe. The term autotoxicity is sometimes used to describe self-attack by the innate immune system so that the term autoimmunity can be reserved to refer to self-attack by the adaptive immune system. In chronic inflammatory situations, both autotoxicity and autoimmunity may occur, either together or as singular processes. Intriguingly, the failure to solve many chronic degenerative diseases may thus reflect a tendency to ignore autotoxicity. Wherever autotoxicity is a major contributor to pathology, the predetermined search for antibodies or offending T cell clones to account for the disease process may distract investigations into the role of the innate immune system.
Indeed, as Elie Metchnikoff pointed out more than a century ago, inflammation starts as a local phenomenon and may remain so. It is therefore pertinent to describe the process in terms of the spectrum of mediators that are generally present at inflamed sites. The biomolecules that characterize inflamed tissue include cytokines, anaphylatoxins, acute phase reactants, proteases, and oxidizers. Many of these molecules are markedly elevated in brain regions affected by Alzheimer's disease and have been reviewed elsewhere (1). We will therefore concentrate on two of the phylogenetically ancient actors of the inflammatory stage—the complement system and the microglia. A primitive system of complement can be traced back to the horseshoe crab, far predating the adaptive immune system. Microglia are related to the phagocytic mesodermal cells of starfish larvae as identified by Metchnikoff.
The Complement System
The classical complement system consists of nine major protein components (i.e., C1, C2, C3…C9) that become serially activated to effect the ultimate destruction of antigenic entities that have been bound by antibody. The cascade of activation reactions begins when the C1 component acquires proteolytic activity as a result of encountering and binding an activator that has traditionally been thought to be an antigen–antibody complex; subunit C1q of the multimeric C1 complex is responsible for this binding (Figure 1⇓). The activated C1 complex cleaves C4 and C2 to produce fragments that associate to form the enzyme (C4b–C2a) that cleaves C3. This modus operandi, whereby the activation of one enzyme gives rise to a second enzymatic activity, effectively amplifies the immunogenic signal. Fragment C3b then associates with C4b–C2a to direct proteolytic cleavage against C5, the event that in turn commits the complement pathway to its terminal, lytic phase. Specifically, fragment C5b causes components C6, C7, and C8 to associate within the lipid bilayer of antigenic surfaces. Finally, multiple molecules of C9 are recruited into this association, where they polymerize into a pore structure, known as the membrane attack complex (MAC), that effects lysis.
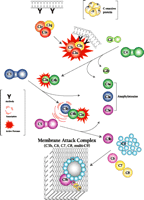
The classical complement system. The nine major complement proteins participate in a series of reactions that involve proteolysis and specific intermolecular associations. Although an immune complex (e.g., antibody bound to the cell membrane of a pathogen, as shown) is the classical instigator of complement activation, C1q can in fact interact with other proteins—some of which are produced in pathological processes—to activate the C1 complex. Many additional factors, such as the C-reactive protein, facilitate the cascade of reactions.
In addition to the direct function of the complement system in mediating the genesis of the MAC, several products of the proteolytic reactions—notably, fragments C4a, C4b, C3a, C3b, and C5a—have important bioactivities within the broader scope of immunity and inflammation. Fragments C4b and C3b can form covalent bonds, in a process called opsonization, with a variety of proteins and other molecules on immune complexes, cells, and bacteria that thereby become irreversibly marked for phagocytic disposal. Fragments C3a, C4a, and C5a, known as anaphylatoxins, induce vascular permeability and stimulate inflammation. Thus, the overall cascade identifies, opsonizes, and destroys its target, while dispatching messengers to seek help.
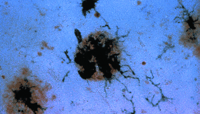
Significantly, all of the complement proteins, including C1q (Figure 2A⇓), exist within the neurofibrillary tangles and neuritic plaques that typify Alzheimer's disease, and C4b and C3b—or fragments derived from them—are particularly prominent (2). Moreover, dystrophic neurites in Alzheimer's disease can be immunostained for MACs (Figure 2B⇓), whereas few or no MACs appear in healthy brain (3). Indeed, there are crucial protective molecules that normally defend host cells against MAC formation so that lysis specifically targets foreign invaders. Nevertheless, if MACs reach a sufficient concentration so as to exhaust the host's autoprotective mechanisms, autologous complement attack can occur. This process, called bystander lysis, is the smoking gun of autodestruction in Alzheimer's disease and leads to the progressive elimination of neurites.
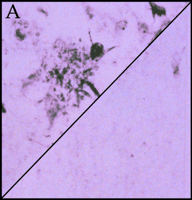
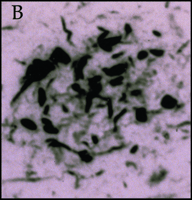
Immunohistochemical staining of the pathological earmarks of Alzheimer's disease.
A. Antibody that is specific for complement protein C1q stains senile plaques in diseased tissue (upper left corner), whereas healthy tissue (lower right corner) is not stained.
B. Dystrophic neurites are stained by antibody that recognizes the membrane attack complex in diseased (but not in normal) tissue.
The discovery of activated complement within the brain raises two important questions. First, how does complement get activated in the absence of antibodies? And second, where do the complement proteins come from? To answer the first question, multiple investigators have shown that three components of senile plaques, namely, the ß-amyloid protein, amyloid P, and C-reactive protein, can bind tightly to C1q to activate the complement system and thereby replace the classical requirement for antibody (4). Most intriguingly, the ß-amyloid protein, amyloid P, and C-reactive protein bind to the collagen-like tail of the C1q molecule, whereas antibody binds to the globular head of C1q. The distinctive mode of activational binding displayed by the senile-plaque proteins is extremely exciting from a therapeutic point of view. Potentially, the autodestructive action of complement might be prohibited by blocking access to the collagen-like tail of C1q. The selective inaccessibility of the collagen-like tail could thereby leave the globular head free for interaction with antibody and thereby reserve activation of the complement cascade for the express purposes of adaptive immunity.
The second question, as to the source of the complement proteins, has been answered primarily by mRNA analysis of brain tissue. It has become clear, through a combination of in situ hybridization and immunohistochemical approaches, that the highest producers of complement proteins are in fact neurons themselves, particularly the cortical pyramidal neurons, which, significantly, are highly vulnerable in Alzheimer's disease (1).
The Microglia
The microglia are the resident phagocytes of the central nervous system. Phagocytes were discovered by Elie Metchnikoff in his great work of 1892. He observed the aggregation of mesodermal cells in starfish larvae that had been impaled with rose thorns. He named the responsive cells phagocytes, and he believed them to represent the main line of immune defense. With respect to the brain, Ramon y Cajal later observed that the central nervous system possesses an “element” in addition to the neuronal and glial cell types. In 1919, Del Rio Hortega revealed this “third element” to be in fact composed of two cell types that he designated oligodendroglia and microglia. He recognized that the oligodendroglia are of epithelial origin, whereas the microglia arise from mesodermal cells that migrate into the brain, where, under the appropriate stimulus, they become phagocytic. The role of the microglia in phagocytosis was challenged into the 1990s, but immunohistochemical studies of brains manifesting Alzheimer's disease, as well as in vitro experiments, have shown microglia to have all the characteristics of phagocytes (5).
Microglia constitute a few percent of the glia of brain and are normally quiescent. When activated by an insult to the brain, they undergo morphological changes and produce massive amounts of oxygen radicals and other materials, which may themselves be toxic to host cells. In addition, immunostaining techniques reveal the induction of many proteins by the microglia, including complement receptors, that are not detected in normal brain tissue. Immunostained microglia appear in clumps on senile plaques (Figure 3⇓) and other sites affected by Alzheimer's disease.
Immunohistochemical staining of activated microglia clustered around a senile plaque. Senile plaques are stained brown with the use of an antibody specific for the ß-amyloid protein. Antibody specific for a membrane protein expressed by activated microglia develop a dark purple color in the staining reaction.
Quantification of the Inflammatory Reaction
Whereas immunohistochemical results suggest an intense inflammatory reaction in brain regions affected by Alzheimer's disease, a more quantitative assessment of the reaction can be obtained by assaying for the mRNAs that encode inflammatory mediators. Fortunately, mRNAs have been found to be surprisingly stable in postmortem tissue, so that they are amenable to extraction and reverse transcription. Subjection of the resulting cDNAs to the polymerase chain reaction yields amplification products whose concentration is proportional to the amount of mRNA originally present in the brain. Figure 4⇓ shows the levels of mRNA encoding each of three mediators of inflammation (i.e., C1q, C9, and C-reactive protein) in the hippocampus of diseased and healthy brain (2). Specifically, the data show that Alzheimer's disease is associated with a marked increase in the levels of transcripts giving rise to complement proteins C1q and C9, as well as of that encoding the C-reactive protein (i.e., an activator of the complement system). The mRNA encoding CD11b (i.e., a complement receptor found on activated microglia) similarly abounds in tissue affected by Alzheimer's disease (E.G. McGeer and P.L. McGeer, unpublished results). The dramatic increase in each of the four transcript levels in Alzheimer's disease, moreover, corresponds to an increase in the level of each mediator as measured by protein gel blotting, and these increases, in turn, show striking specificity for those areas of the brain that are primarily involved in the pathology (2). In areas with a heavy burden of plaques and tangles, such as the entorhinal cortex, hippocampus, and temporal cortex, there are increases of twenty- to eightyfold relative to healthy tissue, whereas only small increases are observed in areas with little pathological involvement, such as the cerebellum. Similar regional differences are found for the mRNAs of other inflammatory mediators studied.
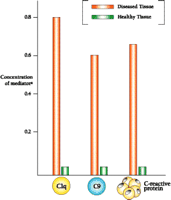
Levels of inflammatory mediators associated with Alzheimer's disease in the hippocampus.
a As determined by mRNA analysis; units are arbitrary and values are normalized.
Therapeutic Possibilities
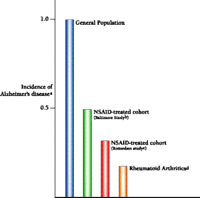
What does the role of inflammation in Alzheimer's disease mean clinically? The first prediction would be that people taking therapeutic measures to ward off traditionally recognized forms of inflammation would be inadvertently protecting themselves against the autotoxic effects of Alzheimer's disease. Indeed, more than twenty epidemiological studies indicate a decreased incidence of Alzheimer's disease in people who are either known to be taking anti-inflammatory agents or are suffering from afflictions in which the administration of anti-inflammatory agents can be assumed (6-11). A particularly compelling study comes from Stewart et al., involving more than 2000 patients who were assessed over extended periods of time in order to determine factors contributing generally to disease (7). The investigators found that the risk of developing Alzheimer's disease was reduced by about one-third for those patients using NSAIDs for two years or less. For those using NSAIDs for more than two years, the risk was reduced by 60 percent. An even greater reduction was recently reported by Veld et al. in studies of chronic NSAID use and was in good agreement with the reduction that we reported in a study of rheumatoid arthritics (Figure 5⇓) (8, 9).
NSAIDs function to inhibit cyclooxygenase (COX) and thus interfere with the production of prostaglandins. Prostaglandins are known to be inflammatory mediators, although they are far less powerful in this regard than the complement system and microglia. NSAIDs thus strike at fringe players in the whole autotoxic and inflammatory scheme. Nevertheless, NSAIDs are well-studied, readily available medications that form a good starting point for approaching anti-inflammatory therapies in Alzheimer's disease. The results obtained with these drugs should encourage the experimental pursuit of agents directed at core activities such as the complement system and activated microglia.
Will NSAIDs work clinically to combat Alzheimer's disease subsequent to an actual diagnosis? A small double-blind clinical trial, led by Joe Rogers in Sun City, indicates that indomethacin may in fact arrest the progression of Alzheimer's disease (10). Another trial, recently reported from Australia, also suggests that diclofenac, a mixed cyclooxegenase inhibitor (i.e., one that targets both the COX-1 and COX-2 isoforms of the enzyme), slowed progression of the disease during the course of six months, although the small size of the study group rendered the statistical significance of these results marginal (11). Trials have also been run with COX-2–selective inhibitors, with reportedly poor results. It should be noted, however, that COX-2 is concentrated in neurons, and several studies have shown that it is involved in synaptic transmission. COX-2–selective inhibitors may therefore not be advised for Alzheimer's therapy. The objective, after all, is to handcuff overaggressive microglia, but not to hammer affected neurons that are struggling to survive and function.
The Innate Immune System in Other Diseases
How about other neurological diseases? Does evidence of an innate immune reaction exist for other neuropathologies? Figure 6A⇓ compares levels of mRNA that encode C9 in areas of the brains from patients with either Parkinson's or Alzheimer's disease. In the case of Parkinson's disease, upregulation is evident in the substantia nigra and caudate, but not in the entorhinal cortex and hippocampus as is seen in Alzheimer's disease (E.G. McGeer and P.L. McGeer, unpublished results). These results are in accord with the disparate regions of brain affected in the two diseases.
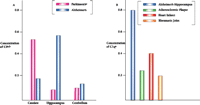
Activation of innate immunity in multiple disease states as indicated by induced levels of complement proteins.
A. Levels of complement protein C9 in three distinct brain regions from patients with Alzheimer's or Parkinson's disease.
B. Levels of complement protein C1q in diseases of disparate organs.
a As determined by mRNA analysis; units are arbitrary and values are normalized.
b Results are similar for analysis of complement protein C1q.
c Levels of C9 in Parkinson's disease are highest in the substantia nigra (data not shown), the primary pathological target.
And what about other organs and tissues that succumb to disease? Do they too have the capacity to produce molecules representative of innate immune system activity? We have examined postmortem tissues from heart damaged by myocardial infarcts (12) and arteries manifesting atherosclerotic plaques (13), as well as biopsies of osteoarthritic tissue (E.G. McGeer and P.L. McGeer, unpublished results). Figure 6B⇑ compares the degree of innate immune system activity, as measured by the levels of mRNAs encoding C1q in Alzheimer-affected hippocampus, infarcted heart, atherosclerotic plaques, and osteoarthritic joints. Interestingly, induction is most pronounced in the case of Alzheimer's disease. And although the bases of the pathologies are quite different—plaques and tangles in Alzheimer's disease, anoxia in heart disease, too much fat in atherosclerosis, and mechanical grinding in osteoarthritis—the secondary process (i.e., activation of innate immunity) is common to all. Thus, the effectiveness of NSAIDs in preventing heart disease and stroke may involve interactions with the innate immune system, in addition to their recognized effects on platelet aggregation.
Conclusions
It indeed appears that chronic inflammation may be a major driving force in the most important diseases of our time: Alzheimer's disease, heart attack, and stroke. We have much to learn about the innate immune system in these conditions. We need to explore the mechanisms whereby the innate immune system recognizes and targets both self and nonself, just as we have investigated these issues in the adaptive immune system. We have every reason to believe that therapies that will selectively modulate the functioning of the innate immune system will prove just as valuable as treatments that intervene with the adaptive immune system.
Acknowledgments
Our work on Alzheimer's disease has been supported by grants from the Jack Brown and Family Alzheimer's Disease Research Fund and the Alzheimer Society of Canada, as well as donations from The Friends of The University of British Columbia, and individual British Columbians.
- © American Society for Pharmacology and Experimental Theraputics 2001
References


