Macromolecular Therapeutics
Emerging Strategies for Drug Discovery in the Postgenome Era
- Department of Pharmacology School of Medicine and Program in Macromolecular Therapeutics University of North Carolina Chapel Hill NC 27599
- Address correspondence to RLJ. E-mail arjay{at}med.unc.edu; fax (919) 966-5640.
Abstract
The postgenome era offers a plethora of potential therapeutic targets. Many of these targets will be addressable using small organic molecules as drug candidates. However, certain aspects of cell function, particularly those that rely on protein–protein or protein–nucleic acid interactions, will be difficult to influence using small molecules. Thus, the possibility of using highly specific macromolecules as potential therapeutic agents is an intriguing concept. Recent developments in several areas of research have brought this possibility closer to fruition. Peptide and nucleic acid combinatorial libraries allow the generation of novel molecules having exquisite selectivity. Structural information and molecular modeling also contribute to the design of new macromolecules with therapeutic potential. Perhaps most importantly, approaches for delivering macromolecules into the cell interior have been developed and applied with considerable success. Thus, the therapeutic use of macromolecules, including oligonucleotides, peptides, and proteins, may be an idea whose time has come.

Introduction
The future of the pharmacological sciences, particularly those aspects relating to drug discovery, has been a much-discussed topic of late. Clearly, the completion of the human genome project will have an enormous impact on future patterns of pharmacological and pharmaceutical research, but exactly how this future will play out remains uncertain. Modern genomics and its offspring, proteomics (1) and gene expression profiling (2), offer an almost overwhelming amount of new information. Much of this information is of an unprecedented nature, and confers such capabilities as the determination of DNA polymorphisms that contribute both to disease and to individual capacities to metabolize and respond to drugs. We are rapidly acquiring new knowledge about the placement of proteins within functional pathways and their patterns of interaction, as well as new information about changes in mRNA levels in response to drugs and disease progression (3). The pharmaceutical industry has rapidly integrated genomic considerations into its research and development programs. For example, target selection, a key aspect of drug development, will greatly benefit from the identification of genes that unambiguously play a role in specific disease processes (4). Likewise, pharmacogenetics (i.e., genetic profiling to predict patient response to a drug) will also be of great value in clinical care (5).

A cell-penetrating peptide effectively delivers a fluorescently labeled oligonucleotide to a cellular interior.
Based on recent progress in the broad realm of genomics, the projected pattern of drug discovery in major pharmaceutical companies seems to be gravitating toward a single model (6). The initial step is target selection based upon genomic or proteomic elucidation of pathways associated with disease. However, as pointed out by Roses (4), the most challenging step may be target validation, that is, reliably defining candidate genes with respect to disease processes. Once a target is identified, it can be rapidly addressed through combinatorial chemistry (7) and high-throughput screening (8). Various screening strategies have been proposed to increase the probability of generating useful leads from chemical libraries, including both focused approaches (8) as well as wider explorations of chemical space and biological pathways (i.e., diversity-oriented synthesis) (9). Finally, lead compounds emerging from the screening laboratory must undergo the tedious process of pharmacodynamic and toxicological profiling before being considered for clinical trials.
The human genome project has revealed approximately 40,000 genes, of which 10,000 or so may be expressed in a given cell at a given time. Thus, there is an incredible diversity of potential drug targets. Nevertheless, the pharmaceutical industry has tended to focus on 10 to 20 percent of the total available targets (8). This subset is rich in G-protein coupled receptors, ligand-gated ion channels, and certain classes of enzymes (e.g., proteases). There are very good reasons for this focus. First, these targets have a history of success in terms of drug development. Second, the ligands for these targets are, for the most part, small organic compounds, which is in accordance with the pharmaceutical “rule of five” (10) that focuses attention on small organic molecules with desirable properties in terms of solubility and membrane permeation (Table 1⇓). Still, the wealth of additional potential targets includes pivotal macromolecules involved in signal transduction, gene transcription, RNA processing, and subcellular organization. Moreover, because macromolecular targets operate primarily by protein–protein or protein–nucleic acid interactions, rather than by binding small organic ligands, they are usually considered to be “undruggable.” Indeed, it has been very difficult to find small organic molecules that can affect critical macromolecular interactions (6, 8). However, if the “rule of five” is set aside, then many more targets are potentially addressable, particularly through the use of peptides, proteins, and nucleic acids as drug candidates.
The idea of using peptides and nucleic acids as drugs has traditionally not been greeted with enthusiasm by the pharmaceutical industry, and there are some very good reasons for this attitude: such molecules are usually unstable in the biological milieu; they are very costly to manufacture; and they often manifest very poor bioavailability. On the other hand, macromolecular drugs possess the potential for exquisite selectivity. Antisense oligonucleotides, for example, can discriminate between mRNA targets differing by only a single base (11). Macromolecules have thus been viewed as pharmacologically recalcitrant but “information-rich,” whereas small organic molecules have by comparison been regarded as tractable but possibly “information-poor.”
In this review, we will outline selected recent developments regarding macromolecules as drug candidates. In that context, we will discuss novel technologies whose impending convergence could radically alter the prospect for using biological macromolecules as effective therapeutic agents. In particular, we will focus on the prospect of using macromolecules that address intracellular targets, thus excluding discussions of well-established approaches such as monoclonal antibodies and recombinant proteins that target the cell surface or extracellular sites.
Macromolecules with Therapeutic Potential
Broadly speaking, there are two general approaches to the creation of macromolecules with therapeutic potential. One approach is to utilize structural knowledge about the target to design a candidate molecule with which it will precisely interact. A simple example of this approach is the design of antisense oligonucleotides that bind specifically to predetermined RNA sequences. The second approach is to use combinatorial, or “evolutionary,” methods, whereby a large library of ligand molecules is screened for affinities and/or specificities with regard to target molecules. The use of peptide or nucleic acid aptamers would exemplify this latter approach.
Oligonucleotides
Antisense Technology
The use of antisense oligonucleotides is perhaps the most mature technology in the development of macromolecular drugs. Base pairing between relatively short oligonucleotides (twelve to eighteen residues) and complementary sequences in mRNA can provide highly selective recognition that can often interdict gene expression. A random sequence of seventeen residues is likely to occur only once in the human genome, and any sequence of thirteen residues would be expected to occur only once in a typical cellular mRNA pool. These probabilities, coupled with the observation that a single base mismatch can result in as much as a 500-fold reduction in binding affinity, underlie the exquisite specificity of the antisense approach (12).
Antisense oligonucleotides can inhibit or modify gene expression via several different mechanisms. The most widely used approach is to trigger the activation of RNase H, the cellular enzyme that degrades the RNA component of RNA/DNA duplexes (12-14). In this scenario, the appropriately designed oligodeoxyribonucleotide will essentially have a catalytic effect in destroying an RNA target (Figure 1A⇓). Another approach is to design oligonucleotides that bind to mRNA at the translation initiation site or at the 5′ cap region so as to prevent translation. Another interesting strategy is the design of oligonucleotides that disrupt RNA secondary structures (commonly, stem-loops) that are necessary for RNA stability. All of these approaches, as well as several others, have been shown to be feasible. The choice of the optimal sequence within the RNA target molecule for annealing the antisense oligonucleotide remains more of an art than a science. Predictions based on simple base-pairing affinity or on computer-generated RNA folding often are not very helpful. The selection of target sequence by “shotgunning” many sites in the RNA target seems to be the current standard (14).
Antisense oligonucleotides can also be used to alter, rather than inhibit, gene expression. In an exciting series of studies, Kole and his colleagues (15, 16), as well as other investigators (17), have shown that antisense oligonucleotides can be used to alter transcript splicing patterns. Approximately 15 percent of genetic diseases are caused by defects in splicing. In many such cases, the correct splice sites exist, but additional, mutant splice sites are also present, resulting in aberrantly spliced mRNA (18). Antisense oligonucleotides directed to the mutant splice sites can force the splicing machinery to use the wild-type splice donor and acceptor sites, thus resulting in correctly spliced mRNA and functional protein (Figure 1B⇓). The most dramatic example of this type of antisense therapy concerns the ex vivo correction of the splicing defect that leads to a form of ß-thalassemia. Specifically, erythroid cells from thalassemic patients were treated with antisense oligonucleotides targeted to aberrant splice sites, thereby resulting in the restoration of hemoglobin A production to 30 percent of wild-type levels (16). This promising result suggests that a wide variety of other genetic diseases that involve aberrant splicing might also be susceptible to antisense-based correction.
Recent developments in the biological application of antisense technology have been based on important advances in oligonucleotide chemistry (19, 20). Naturally occurring oligodeoxyribonucleotides having phosphodiester linkages are far too unstable to be used in vivo. Thus, a variety of chemically modified oligonucleotides have been developed. Most widely used are the phosphorothioates, in which a sulfur atom replaces one of the oxygen atoms in the diester linkage. This modification increases stability to nucleases; however, phosphorothioates retain the DNA-like ability to recruit RNase H. Another common approach is to derivatize the 2′ position of nucleotides in order to synthesize oligonucleotides that better recognize their RNA target and are more resistant to nuclease attack; these properties, however, are accompanied by an inability to mobilize RNase H. Several classes of oligonucleotides with uncharged backbones have also been synthesized, including the methylphosphonates, the peptide-nucleic acids, and morpholino derivatives. The latter two types of oligonucleotides can have very high affinities for RNA targets and very high nuclease resistance, but are again unable to recruit RNase H. An interesting recent approach has been to synthesize oligonucleotides of very stable (e.g., 2′-O-methoxy-derivitized) residues, which contain a limited internal sequence (i.e., a “gapmer”) of six to eight phosphorothioates that, although relatively unstable, renders its target labile to attack by RNase H. This “best-of-both-worlds” approach is sometimes quite successful (21).
Over the last several years, antisense technology has begun the move from the laboratory to the clinic (12). Vitravene (ISIS Pharmaceuticals), for the specialized treatment of viral retinitis, is the first antisense drug to reach the market, and several other agents are in phase I or II clinical trials. Cancer-related targets such as Bcl-2, Raf-1, the PKC isozymes, and BCR-Abl have attracted a good deal of attention, and targets involved in inflammatory (ICAM-1) and viral diseases are also being pursued (13, 22).
A recent article by Gewirtz (23) illustrates both the promise and the tribulations of attempts to treat cancer with antisense agents. On the whole, antisense drugs have shown some promise in clinical trials, but not as much as had been hoped. There are, however, several likely reasons for this initial disappointment. First, all of the early clinical trials have been conducted with phosphorothioate oligonucleotides. These compounds bind extensively to proteins and thus cause many biological effects (side effects) unrelated to their antisense action. Second, as might be expected from their polyanionic character, most antisense oligonucleotides are very poor at crossing cell membranes and result in less than ideal biodistribution (24). Thus, relatively large doses of antisense compounds must often be given to obtain modest intracellular levels. Interestingly, however, whereas in the absence of a delivery agent there is virtually no uptake of phosphorothioate oligonucleotides by cells in culture, there is clear evidence of uptake by cells in vivo, indicating important differences in oligonucleotide transport in the two situations (13). Further, as indicated above, advances in oligonucleotide chemistry may ameliorate clinical responsiveness. New chemical syntheses have resulted in antisense compounds that are much more potent than the phosphorothioates and display less non-specific binding to cellular constituents. In terms of delivery and uptake problems, some of the newer derivatives, particularly the morpholino compounds, display better membrane penetration (25). In addition, new technologies are evolving that will allow the effective intracellular delivery of a variety of large molecules, including antisense oligonucleotides (see below). Thus, despite early setbacks, antisense technology will likely play an important role in therapeutics.
Triplex-Mediated Efficacy
Triplex-forming oligonucleotides, chemically similar to antisense oligonucleotides, associate with double-stranded DNA through non-Watson-Crick base pairing (i.e., Hoogsteen or reverse-Hoogsteen bonding) that generally depends on polypurine-rich tracts. Several forms of triple helices are possible, and various intercalating agents or chemical cross-linkers can be used to enhance formation of the triplex structure (26). One of the original goals of triplex research was to inhibit gene expression by blocking transcription, ostensibly by displacement of transcription factors, interference with the recruitment of RNA polymerase, or inhibition of transcriptional elongation (Figure 1C⇓). Although there have been some clear examples of influencing transcription through triplex-forming oligonucleotides, this strategy has been hampered by problems with both cell penetration and access to the densely packed nuclear DNA. Recently, however, another very interesting strategy has emerged that is compatible with the relatively low efficiency, but high selectivity, of triplex formation in living cells. Specifically, triplex-forming oligonucleotides have been used to create targeted mutations in genes. Triplex formation dramatically increases the frequency of mutations at the target site, probably by a nucleotide excision repair mechanism. This strategy has been used to introduce mutations into a gene in mice (27), which raises the interesting prospect of gene therapy to correct genetic abnormalities arising from point mutations.
Ribozymes and DNAzymes
Since the initial discovery of self-splicing RNA twenty years ago, there has been intense interest in the mechanisms, biological significance, and possible practical applications of catalytic nucleic acids (14, 28, 29). Most work has centered on ribozymes, short RNA molecules that can catalyze sequence-specific cleavage of target RNA molecules. The specificity is provided by recognition sequences that flank the catalytically active motif. Several RNA catalytic domains have been identified, but the most common are the hammerhead and hairpin structures from viral genomes. Although catalytic activity is dependent on core sequence motifs, the flanking sequences may undergo chemical modification without loss of cleaving activity (29). Thus, a variety of ribozymes with increased stability in the biological milieu have been synthesized. The cellular uptake and biodistribution of chemically stabilized ribozymes seems to be similar to that of antisense oligonucleotides of comparable size (30). As with antisense compounds, the uptake of ribozymes is more effective in vivo than in cell culture (29). Ribozymes can be encoded within vectors and subsequently expressed within the cell (29).
An interesting new development is the advent of catalytic DNA, or DNAzymes. For example, the “10-23” RNA-cleaving DNAzyme offers a high catalytic efficiency coupled with great specificity and inherently greater stability than a typical ribozyme (28). Although ribozymes and DNAzymes are very interesting from a number of viewpoints, their application to pharmacological problems has lagged behind antisense approaches, likely because of the greater inherent complexity of catalytic nucleic acids. Nonetheless, a number of studies of ribozyme action in vivo have been reported, and three early-phase clinical trials are currently taking place (29).
Aptamers: Combinatorial Synthesis of Binding Activities
Although many scientists usually think of peptides or oligonucleotides as linear sequences of amino acids or bases, such molecules in solution form stably folded structures. Peptide and nucleic acid aptamers are molecules, with specific three-dimensional configurations, that are isolated from large oligomer libraries using powerful physical or genetic selection techniques. Aptamers can exhibit exquisite specificity for their intended targets and can often be selected rather rapidly. However, at present their potential use in therapeutics is constrained by issues of stability and biodistribution.
Nucleic Acid Aptamers
Nucleic acid aptamers, either RNA or DNA based, are usually isolated by a version of the SELEX (for systematic evolution of ligands by exponential enrichment) technique originated by Gold and colleagues (31). A combinatorial library usually consists of randomized sequences, twenty to forty nucleotides long, that are flanked by specific sequences that allow for amplification by the polymerase chain reaction (PCR). The synthesis of all possible single-stranded nucleic acids forty residues in length from a pool of four nucleotides gives rise to 440 (i.e., circa 1024) distinct sequences; such a library of molecules will thus fold into a great variety of configurations. To screen for candidate molecules with specific affinities, the library is exposed, so as to promote binding, to a target (i.e., a protein, nucleic acid, or small molecule). Complexes of candidate molecule and target can then be recovered by affinity purification and subjected to PCR (Figure 2⇓). Successive rounds of affinity screening, or “selection,” can lead, in a Darwinian manner, to aptamers with very high binding affinities (32). Indeed, the affinities of RNA aptamers for their targets is often substantially greater than those of immunoglobulin Fab fragments for their corresponding antigens. In addition, aptamers are capable of exquisite specificity (33). For example, an RNA aptamer has been described that binds to theophylline with a 10,000-fold greater affinity than to the related compound caffeine.
Recently, careful physical studies, including x-ray analysis of several complexes of nucleic acid aptamers with their protein targets, have led to important insights into the affinity and specificity of aptamer interactions. It seems that the enclosure of large portions of the target by the nucleic acid contributes to both affinity and specificity. Unlike proteins, which are built from an array of twenty chemically varied amino acids, any given nucleic acid aptamer must derive its distinct chemical properties from a mere four nucleotide building blocks. Nucleic acid aptamers can thus fail to offer the versatility of surface shape and charge that are available in protein–protein interactions. The rather limited chemical functionality on which any nucleic acid aptamer can depend in interacting with its ligand is compensated, however, by maximizing contact through deep enclosure of the ligand (33).
Because native RNA and single-stranded DNA are generally too labile for therapeutic applications, nucleic acid aptamers need to be optimized for stability in biological fluids, as well as for more desirable pharmacokinetics and biodistribution. As with antisense technologies, the development of aptamers is benefiting from a great deal of novel nucleic acid chemistry. Thus, modifications at the 2′ position of ribose moieties and at the 3′ terminus can increase resistance to nucleases (32); however, the determination of which chemical modifications can be made without disturbing the desirable binding properties of the aptamer is not trivial.
To date, most of the intended therapeutic targets of nucleic acid aptamers have been extracellular, such as elements of the blood clotting cascade (e.g., thrombin) (32). However, it seems likely that nucleic acid aptamers will take advantage of the intracellular delivery technologies currently being applied to antisense oligonucleotides and therapeutic peptides (see below), so that aptamers could also target cytosolic or nuclear targets. One of the interesting characteristics of nucleic acid aptamers and antisense oligonucleotides is that they, unlike peptides and proteins, do not seem to elicit an immune response. Such immune tolerance could be an essential aspect of future therapeutic development.
Peptide Aptamers
Peptide aptamers have also engendered considerable interest. Here the strategy is to identify relatively short peptide sequences that bind selectively to predefined targets. One important approach has been to use phage display (34, 35), whereby libraries of peptides are fused to phage coat proteins. Another approach has been based on the yeast two-hybrid technique, in which a constrained peptide library is displayed in the context of a surface loop of a larger protein (e.g., E. coli thioredoxin) and screened for interactions with targets expressed in the cell. In the most straightforward application of the latter strategy, the peptide library is screened in yeast versus a known target protein. For example, an early study used this approach to screen a series of peptides and identify nanomolar affinities—in some cases proving inhibitory—for Cdk2 kinase (36).
Another application of peptide aptamers has been as affinity reagents to identify both known and novel key players of biological pathways by “knocking out” elements, for example, in developmental pathways of Drosophila (37); cell cycle checkpoints have similarly been studied in yeast (38, 39). Unfortunately, peptide aptamers have not yet progressed very far toward pharmacological and therapeutic application. One hurdle to effective peptide aptamer therapy will be in developing peptide libraries that mimic the full array of structures represented in the cellular proteome. The imposition of chemical linkages, such as disulfide bridges, may provide additional control in developing peptide libraries. Nonetheless, the peptide aptamer approach provides a powerful strategy that may lead either directly to the identification of therapeutic peptides, or to the provision of lead molecules that can serve as the basis for the design of peptidomimetics.
“Designed” Proteins: Selective Regulation of Gene Expression
A strategy that is of great interest to our group is the use of combinatorial libraries to create “designed” proteins able to modulate various biological events. In particular, we, and others, have addressed the issue of controlling gene expression with such designed proteins. The basic idea is to use the modular nature of transcriptional regulators to create chimeric molecules composed of a transactivating or repressor domain fused to a promoter-specific, DNA-binding domain. The specific aim is to attain selective activation (or repression) of a specifically targeted gene. Significantly, this goal requires a much more precise capability for DNA recognition than is normally found in native transcription factors that must often regulate multiple genes or gene families.
The evolution of this approach began in the early 1990s with the convergence of two important concepts. The first was that peptide or protein libraries could be “displayed” on the surface of filamentous phage, and that such “phage display” could provide a powerful technique for the selection of molecules with strong and specific binding affinities to virtually any sort of target (34, 40, 41). The second was the realization that zinc finger (Zif) DNA-binding domains function in essentially a modular fashion (42); one could thus think about mixing Zif domains from different members of the large Zif family of transcription factors, or even about selecting novel Zifs with desirable DNA-binding specificities. Several groups therefore proceeded to apply phage display to select novel Zifs with altered cognate DNA specificity (43-45).
Recent approaches to develop transcriptional regulators with novel DNA-binding characteristics provide an interesting perspective on some of the issues involved in trying to develop macromolecules as potential therapeutic agents. To follow this story it is necessary to review some basic information concerning the family of Zif transcription factors and their interactions with DNA. In one subgroup of the family, each repeated Zif consists of approximately thirty residues, including two cysteines and two histidines that coordinate a zinc ion and thereby stabilize both ß-sheet and α-helical regions within the “C2H2” Zif domain (42). DNA-binding interactions occur between bases and the α-helical region, and between the phosphate backbone via ß-sheet residues.
To a first approximation, each C2H2 Zif recognizes a single DNA triplet. It is because of this modular nature that individual Zifs can be combined to generate new DNA-binding specificities (45, 46). However, this attractive scenario is limited by certain complexities. Each C2H2 Zif, for example, establishes contact with the DNA strand complementary to its own cognate triplet (47, 48), and subtleties within the linker sequences between adjacent Zifs are important (49-51). Finally, the Zif entities that have been used thus far as backbones for phage display show a strong preference for GC-rich motifs, making it difficult to target AT-rich promoter sites (52). Thus, only some of the interactions between C2H2 Zifs and DNA can be rationalized in terms of a generalized “triplet recognition code” (48, 53).
An important limitation is that phage display is applicable only for protein–DNA interactions in vitro. The first transcription factors to be “designed” from Zif modules bound well to their intended targets in vitro, but their use to regulate transcription in reporter gene assays was initially somewhat disappointing (54). More recently, however, Zifs selected by phage display have been incorporated into transactivators and repressors that function reasonably well in reporter gene assays (54, 55). However, the episomal reporter constructs that have been used differ from the organization of chromosomal DNA. Thus, an important question was whether “designed” transcription factors would be able to influence expression of an endogenous chromosomal gene. Studies from our laboratory using a yeast library have validated the approach for the multidrug resistance MDR1 gene (50), as has work from other laboratories for the erbB-2 gene (56) and a stably integrated reporter gene (57) using phage display.
Some of the disadvantage of in vitro screening by phage display may be overcome by yeast-based methods (58). This approach screens for protein–DNA recognition under physiological conditions where the DNA target is part of a chromosomal structure. There is a drawback to this approach, however, in that yeast libraries are inherently much less complex than phage libraries. A recent development that partially circumvents the relative simplicity of yeast libraries is a technique for screening protein–DNA interactions in bacteria (59). Another interesting development is the “design” of transcription factors that bind to their DNA cognate sequences in response to small molecules. In this context, a DNA-binding module containing six Zifs was linked to modified steroid-binding domains of hormone receptors. The resulting chimeric transcription factors specifically interacted with 18-base target sites in response to either 4-hydroxytamoxifen or RU486, depending on the given construct (60). Thus, several new approaches are coming into play and should permit the efficient creation of novel, selective, regulated transcriptional activators and repressors.
In addition to combinatorial libraries, computer-assisted molecular design has received considerable emphasis for the creation novel DNA-binding proteins. This approach requires x-ray or other high-quality structural information to guide the design of chimeric proteins with DNA-binding domains. For example, molecular modeling aided the design of a DNA-binding domain comprising three Zifs linked to the C terminus of a TATA-binding protein (54). A similar approach was used to link specific Zifs along with the dimerization domain of the yeast transcription factor GAL4, resulting in a dimeric fusion polypeptide capable of highly specific DNA binding (61). Molecular modeling has also been applied to the assembly of proteins from multiple Zif modules initially selected by phage display (51). In our laboratory we have recently used computer modeling to create chimeras between Zifs and the DNA-binding domain of p53 (Figure 3⇓). This technique has allowed us to prepare transcriptional regulators that affect subsets of genes downstream from p53 (unpublished results). Thus, molecular modeling can complement library search strategies in the creation of novel DNA-binding domains.
Intracellular Delivery ofLarge Molecules
A key issue for the future evolution of macromolecular therapeutics is the formidable problem of effectively delivering large molecules to the interior of cells. Cell membranes exquisitely regulate the permeation of small molecules such as sugars and ions and, in general, are completely impermeable to large polar molecules such as proteins and nucleic acids. However, there are several examples of macromolecules that can efficiently cross membranes and enter cells. Many viruses have evolved ways to deliver both viral proteins and nucleic acids to the cytoplasm and nucleus (62), and certain bacterial toxins also can traverse the cell membrane (63). Another very interesting example concerns several transcriptional regulators, including the human immunodeficiency virus (HIV) Tat protein, and the Antennapedia, Engrailed, and Hoxa-5 homeodomain proteins from Drosophila; in fact, these proteins can apparently migrate from one cell to another (64). Recently, investigators have borrowed concepts and tools from such examples, and have devised a variety of chimeric macromolecules capable of entering cells.
An important new strategy for large-molecule therapeutics involves so-called “delivery peptides,” or “cell-penetrating peptides,” that can be less than a dozen amino acids long (65, 66). Examples include a polybasic sequence from the HIV Tat protein (66), a polybasic sequence from the Drosophila Antennapedia protein (65), and amphipathic sequences based on signal peptides (Table 2⇓) (67). These cell-penetrating peptides can be coupled to other peptides (68) or oligonucleotides (69), thus facilitating the entry of otherwise non-penetrating molecules into cells. Our laboratory has recently used polycationic Tat- and Antennapedia-derived peptides to deliver antisense oligonucleotides that target the nuclear MDR1 gene (70). Other examples include the use of amphipathic, signal peptide–derived sequences to deliver inhibitory peptides that selectively interfere with G-protein coupling (71) or nuclear translocation of transcription factors (72).
In a very impressive series of studies, Dowdy and colleagues have used a Tat-derived peptide to deliver a variety of large proteins to intracellular locations (66). Chimeras were expressed in bacteria, purified, and then added externally to mammalian cells that consequently underwent biological programs (e.g., signal transduction, cell cycle regulation, apoptosis) consistent with the effective delivery of the given protein (73-76). Similar chimeras may well function in vivo, as indicated by a recent report that ß-galactosidase can be delivered across the blood brain barrier (77). Along the same conceptual lines, a 12-residue hydrophobic peptide was used to deliver a chimeric protein containing the Src homology 2 (SH2) domain into cells so as to disrupt protein–protein interactions essential to the activation of mitogen-activated protein kinase (78).
Relatively large protein domains can also prove to be effective as delivery modules. The VP22 protein from herpes simplex virus has been shown to traffic between cells. Plasmids or retroviruses constructed so as to express chimeric proteins that combine the VP22 sequence with a functional (e.g., enzymic) protein of choice can enable transfected/transformed cells to traffic functional chimeras into the cytoplasm and nuclei of neighboring cells (79-82). Although VP22 may be very interesting from the perspective of gene therapy, the data supporting uptake of purified VP22 chimeric proteins from the extracellular compartment is rather limited, and it is possible that cell-to-cell contact may be needed for VP22 based delivery. The membrane translocating domains (83) of bacterial toxins have also been used for the intracellular delivery of chimeric proteins (84). In a recent example, the Bcl-xL apoptosis inhibitor was successfully delivered to cells as a fusion protein with a diphtheria toxin domain (85).
The mechanism underlying delivery by cationic cell-penetrating peptides remains somewhat mysterious (65, 66). The delivery appears to involve neither specific receptors nor endocytosis, but rather occurs directly across the plasma membrane. Two interesting models have been proposed for the action of polycationic delivery peptides. One model suggests that, as the peptides bind to anionic sites in the lipid bilayer, they cause a transient membrane destabilization, possibly involving non-bilayer structures (65). The delivery peptide and its fusion partner would then cross the membrane at these disordered sites. An alternative model suggests that the cationic delivery peptide binds to the membrane and that the fusion partner “wiggles” across the membrane in denatured form, followed by chaperone-mediated folding within the cytoplasm (66).
Neither of these models is supported by a substantial body of biochemical or biophysical evidence, and the requirements of protein unfolding and refolding in the protein delivery process require particular investigation. Another issue concerns the delivery of cargo to specific intracellular compartments. However, some interesting studies are starting to emerge on the mechanisms whereby membranes interact with polybasic delivery peptides and VP22 (86-88). And as pointed out elsewhere (66), there is no a priori reason to expect that all cell-penetrating peptides will utilize the same mechanism. It is possible that insights will be garnered from the body of literature concerning biophysical aspects of interactions between phospholipids and peptides (89) as well as between toxin subunits and membranes. One of the interesting concepts to emerge from the latter work is the “molten globule” as a partially denatured state that allows effective interactions with membranes (83). Finally, with rare exceptions, virtually nothing has been done in terms of understanding the in vivo behavior of chimeric macromolecules linked to delivery peptides. Certainly, issues concerning pharmacokinetics, biodistribution, and immunogenicity will need to be thoroughly explored before any pharmaceutical applications become feasible.
Cellular Targeting
The ability to target macromolecules to a specific cell type has obvious therapeutic consequences. There is a long history of research in cell targeting, largely stemming from the monoclonal antibody and liposome fields. (90). Here, we briefly mention some exciting new developments involving the use of combinatorial library approaches to create cell-selective reagents.
Phage display can be used to identify polypeptides that bind to those surface receptors that are characteristic of a particular cell type (91). Although this approach has been used for cells in culture, a pioneering study by Pasqualini and Ruoslahti (92) demonstrated that phage selection in vivo can result in tissue-specific binding. Specifically, this approach led to constrained peptides that selectively target angiogenic endothelial cells. Peptides have been coupled to conventional antitumor drugs (93) and cytotoxic peptide sequences (94), resulting in significant anticancer activity in animal models. In principle, this technology makes it readily possible to identify relatively small peptide ligands that can bind preferentially to any desired cellular target. Such peptides could be coupled to therapeutic oligonucleotides, peptides, or proteins, thereby providing targeted macromolecular pharmaceutical agents.
Perspective
Over the last few years there has been a substantial upsurge in research on possible therapeutic applications of large molecules. Antisense technology has progressed the furthest, with several antisense oligonucleotides now in advanced clinical trials and one on the market. Other approaches, including ribozymes, triplex-forming oligonucleotides, aptamers, and designed proteins are at earlier stages of development. It is clear that most of these approaches are capable of providing highly selective recognition of target genes or proteins.
Nevertheless, many issues remain to be addressed before therapeutic macromolecules can become a practical reality. First, there is the question of efficacy. Whereas many of these approaches may work in model systems, their ability to function effectively in a practical context is unclear. We have sought, for example, to inhibit the expression of P-glycoprotein, the product of the MDR1 gene that is responsible for a multidrug resistance phenotype in cancer cells. We have utilized both antisense approaches (70) and “designed” repressor proteins (50) and have attained clear-cut effects. However, our best efforts to date have produced no more than a 70 to 80 percent reduction in P-glycoprotein expression, which simply is not good enough. To attain clinically useful effects, we would need to reach 90 percent inhibition of expression, at the very least. It seems likely that many other therapeutic targets will similarly demand such stringent standards that exceed the capabilities of current macromolecular technologies.
Another key question involves the delivery of potentially therapeutic macromolecules. Although some of the types of molecules discussed above (e.g., ribozymes, aptamers, “designed” transcription factors) could be generated intracellularly by means of gene therapy approaches, the direct delivery of the active molecule seems much more appealing. The dramatic recent results of Dowdy and colleagues using the Tat peptide to deliver proteins, as well as many other studies using cell-penetrating peptides to deliver oligonucleotides or other peptides, suggest that major inroads are being made into the problem of intracellular delivery. Nevertheless, the in vivo biodistribution of proteins and nucleic acids linked to Tat or other delivery peptides is almost completely uncharacterized.
Finally, the interactions of novel therapeutic macromolecules with the immune system must be considered. Although there have been few problems reported thus far with immunogenicity in animal or clinical studies using antisense oligonucleotides and ribozymes, it is clear that nucleic acids can have strong impacts on the immune system (95). In the case of peptides and proteins, the immunological concerns are very clear. Novel peptide sequences, regardless of whether they are designed for purposes of function or delivery, will likely trigger strong immunological responses. It is thus possible that such molecules will have to be restricted for use in immune-compromised individuals. In summary, the exciting prospect of developing macromolecules as therapeutic agents will pose formidable challenges. Nevertheless, the potential rewards of highly specific, or “information-rich,” macromolecular therapeutics make such challenges inviting.
The Rule of Five
Cell-Penetrating Peptides Under Investigation
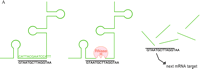
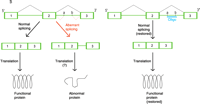
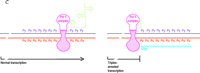
Mechanisms of oligonucleotide therapy.
A. RNase-H–mediated degradation of antisense-targeted mRNA. An antisense oligonucleotide (shown in black) that mimics a deoxyribose-phosphate backbone base pairs with a specific sequence in an unstructured region of an mRNA (green) target (left). The resulting RNA/DNA-like heteroduplex (center) is a suitable substrate for RNase H (red), which then cleaves the mRNA and opens the way to further degradation by other cellular nucleases (right). The oligonucleotide is released and cycles to other molecules of the mRNA target.
B. Inhibition of cryptic splice sites. Mutations that result in cryptic splice sites (denoted as a and b) can cause aberrant processing (indicated in red) of pre-mRNA (green) so that nonfunctional protein is produced (left). Therapeutic use of an RNA-type oligonucleotide (oligo; blue) blocks the cryptic splice site and restores normal splicing so that normal mRNA and functional protein are produced (right).
C. Triplex inhibition of transcription. The RNA polymerase (pink) complex is normally processive (left), moving along double-stranded gene sequences (purple and red) to produce mRNA (green). The presence of a triplex-forming oligonucleotide (oligo; blue) in a purine-rich tract (right; Pu, purine; Py, pyrimidine) interrupts the processivity of the polymerase complex and thus interdicts transcription (right).
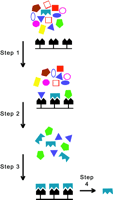
Selection of aptamers in vitro.
A library of nucleic acid aptamers is created by the SELEX method. The initially crude library of synthetic oligonucleotides (colored geometries) is exposed to the target molecule (black) bound to a surface (prior to Step 1). Members of the library that do not recognize the target are not retained, whereas bound members are “selected” into a residual library (result of Step 1). The residual library is amplified by PCR (Step 2), and further rounds of selection refine the aptamer pool (Step 3), ultimately purifying the optimal ligand (Step 4).
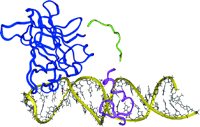
Computer-assisted design of a novel transcription factor for the BAX gene.
The BAX promoter contains sites for the binding of both p53 and of zinc finger transcription factors. The structure of the DNA-binding domain of p53 (blue) and of a zinc finger from the SP1 transcription factor (purple) were obtained from protein structure data banks. The protein domains were docked onto double-stranded DNA (yellow) using a molecular modeling program. A polypeptide linker (green) was designed to optimally position both DNA-binding domains onto the promoter. A transactivating domain was also added (not shown). The information from modeling was incorporated into the design of a vector to express the chimeric protein (see text for details; unpublished work).
Acknowledgments
Work from the authors' laboratory was supported by grants PO1 GM 59299 and RO1 CA 77340 to RLJ. The authors would like to thank their colleagues in the UNC Macromolecular Therapeutics Program, M. Cho, R. Kole, G. Pollack, D. Thakker, and R. Samulski, for their encouragement and support. The authors also thank Brenda Asam for superb editorial assistance.
- © American Society for Pharmacology and Experimental Theraputics 2001

