Spinal Astrocytes in Pain Processing: Non-Neuronal Cells as Therapeutic Targets
Abstract
The treatment of chronic inflammatory and neuropathic pain is a major concern, and the need for new more effective analgesics with less adverse effects is immense. Traditionally, mechanisms proposed for pain modulation have centered almost exclusively on peripheral or central neurons in the pain pathways. Recent research, however, indicates that activation of microglia and astrocytes in the dorsal horn of the spinal cord is of central importance for the development of chronic pain states. Understanding the intercellular communication among astrocytes, microglia, and neurons in the dorsal horn during acute and chronic pain may be instrumental for the development of new analgesic drugs. The purpose of this review is to highlight the role of astrocytes in regulating pain processing.
Introduction
The ability to perceive pain allows organisms to respond to imminent danger and thereby minimize injury. In many pathological conditions, however, pain has lost its protective role and may instead cause substantial suffering. Clinically, the treatment of pain is a major challenge, and the need for new, more effective analgesics with less adverse effects, especially with regard to the management of chronic pain, is immense. Traditionally, pain has been thought of as an alarm function mediated almost exclusively by neurons. More recently, non-neuronal glial cells in the central nervous system (CNS) have also been implicated in various animal models of chronic pain. Intricate interactions between neurons and glial cells in the dorsal horn of the spinal cord are evident under both normal and pathological conditions. In response to nerve trauma and inflammation, sustained activity in primary afferent nociceptive (pain) neurons appears to activate glial cells in the dorsal horn and provide a common etiological mechanism for pain in a number of pathological conditions.
There are three main types of glial cells in the central nervous system: microglia, astrocytes, and oligodendrocytes. Of these, investigation into chronic pain has tended to focus upon the microglia. However, there is growing evidence that astrocytes are also critical for promoting and maintaining chronic pain conditions. The purpose of this review is to summarize the current knowledge about the role of astrocytes in chronic pain and to highlight some of the molecular mechanisms that have been suggested to mediate the intercellular communication between astrocytes and neurons in the dorsal horn during acute and chronic pain.
Pain Signaling
To designate sensation of a stimulus that has the potential to cause tissue injury, Sherrington used the term “nociception” (from the Latin nocere, to harm) (1). This type of perception is typically self-limiting; for example, touching a hot object forces rapid withdrawal of contact, and if there is no injury, the sensation of pain dissipates quickly. Although the magnitude of the stimulus necessary to evoke a “criterion escape behavior” or a verbal response is generally high, this nociceptive threshold can be shifted in nerve injury and inflammation. A normally non-noxious (i.e., non-painful) stimulus or a mildly aversive stimulus that evokes pain-related behavior is referred to as allodynia or hyperalgesia, respectively.
Most tissues in the body are innervated with nociceptors, which are the free endings of small-diameter primary afferent neurons (sensory neurons) that are activated by stimuli associated with tissue damage. Nociceptors respond to noxious thermal or chemical stimuli through the activation of membrane transducer proteins known as transient receptor potential (TRP) receptors (2). Compared to the myelinated (Aα and Aβ) fibers, whcih respond to non-noxious stimuli (e.g., light, touch, and vibration), the nociceptive neurons typify slower-conducting, thinner, and only lightly myelinated (Aδ) or unmyelinated (C) fibers.
Nociceptive sensory neurons synthesize and release, in response to noxious stimuli, a number of neurotransmitters and neuromodulators from their terminals within the dorsal horn. These released substances include glutamate, substance P (SP), calcitonin gene related peptide (CGRP), adenosine triphosphate (ATP), brain-derived neurotrophic factor (BDNF), somatostatin, vasoactive intestinal polypeptide (VIP), galanin, and vasopressin. In addition, nociceptive signaling is modulated by interactions in the dorsal horn between terminals of sensory neurons, ascending projection neurons, interneurons, and terminals of brainstem neurons forming descending pathways. Spinal interneurons may be excitatory or inhibitory (containing, for example, gamma-amin-obutyric acid (GABA), glycine, or opioid peptides). Of importance for this review is the observation that the nociceptive signal is also modulated by interactions between neurons and glial cells in the dorsal spinal cord.
In the dorsal horn, nociceptive signals are relayed to projection neurons with axons that cross the midline and ascend, on the contralateral side of the spinal cord, to thalamic nuclei (Figure 1). As these projections ascend, a large number of fibers branch off to synapse in various regions in the brainstem and midbrain, including the rostral ventromedial medulla (RVM), locus coeruleus, and the periaqual gray (PAG). From the thalamus, tertiary neurons convey the nociceptive message to discrete regions of the cerebral cortex, where the sensation of pain, with its sensory discriminative and affective components, is formed. Descending pathways, with cell bodies in the RVM, the locus coeruleus, and the PAG, can either inhibit or facilitate nociceptive signaling via release of norepinephrine, serotonin, and other neurotransmitters in the dorsal horn.
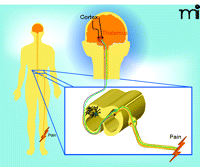
Nociceptive signaling. C fibers (non-myleinated) and Aδ fibers (lightly myelinated) are schematized in green and orange, respectively. Astrocytes and microglia (green and white, respectively, here shown on the contralateral side) have been implicated in the modulation of nociceptive signals. Several of the neurotransmitters and neuromodulators that mediate communication between neurons and non-neuronal cells in the dorsal horn have been identified. See text for details.
During peripheral tissue damage, injured tissue and infiltrating inflammatory cells release a number of factors that reduce the threshold for activation of peripheral sensory neurons. Factors involved in this process of “peripheral sensitization” include tumor necrosis factor (TNF), interleukin 1β (IL-1β), prostaglandins (PG), bradykinin, nitric oxide (NO), ATP, and neurotrophins. There is a concurrent release of such mediators in the spinal dorsal horn, which facilitates pain perception by amplifying the nociceptive impulse conveyed to the brain (3, 4). This activity-dependent facilitation, termed “spinal sensitization,” is mediated by the release of glutamate, SP, NO, and PGs, among other factors. Neurocentric mechanisms that have been proposed to account for spinal sensitization include long term potentiation (LTP) in dorsal horn neurons (5), increases in peptide content, alterations in the expression of peptide receptors and sodium channels in sensory neurons (6), altered AMPA receptor expression and trafficking in dorsal horn neurons, increased facilitatory tone in descending pain modulating pathways (7), and disinhibition in descending pathways or intrinsic neurons at segmental levels (4).
Intriguingly, recent studies implicate the activation of glial cells in the dorsal horn as a mechanism of spinal sensitization. For example, the release of BDNF from microglia, which results in an increased glutaminergic and decreased GABAergic transmission in the dorsal horn, seems to be important in increasing pain perception (8–10). As was first shown by Garrison and collaborators (11), activation of astrocytes also appears to be a mechanism for persistent pain (see below). It is important to note, however, that there are a number of etiologies for chronic pain and a variety of experimental models in pain research. Thus, the pattern of changes in the dorsal horn may differ markedly among experimental settings. It is also impotant to note that while there is evidence for an important role of glia in animal models of pain, direct evidence for a role of central glia in chronic human pain states is still lacking (12).
The Glia: Surrounded in Prejudice
Although they far outnumber neurons in the central nervous system, the role of glial cells in regulating neuronal function has been poorly defined, and they have been relegated to a “housekeeper” status. This limited view of glial function was supported by i) early morphological comparisons of glia s to the connective tissue cells that “support” muscles and glands; ii) the lack of synaptic contacts between glia and neurons; and iii) electrophysiological determination that glia have a stable membrane potential (13). By the end of the seventeenth century, however, Ramón y Cajal recognized that glia might play a functional role that was not reflected by cell morphology (14). Fifty years ago, Robert Galambos introduced the concept of gliapses (e.g., glia–neuronal contacts) (15) through which glia and neurons could function in concert. However, Galambos did not discern the three main classes of glial cells, and so the role of astrocytes in encapsulating synapses and mediating neuronal–glial signaling was overlooked (16). In addition, it was only recently demonstrated that although microglial cells may seem rather quiescent under basal conditions, they perform important immune surveillance and are readily activated upon injury (17). Microglial cells are increasingly recognized as part of the functional unit of the synapse, contributing to the generation and maintenance of excessive excitatory transmission. One may therefore construe a tetrapartite synapse, which includes astrocyte, microglia, and the pre- and post-synaptic neuronal terminals (12, 18, 19).
Astrocytes
Whereas microglia have been described as the “first line of defense” and “the resident macrophages of the central nervous system”, astrocytes have been viewed as “gap fillers” and “housekeeping cells,” providing structural and nutritional support, maintaining proper extracellular ion, pH, and glucose levels, and scavenging neuronal waste products (20). This conservative view of astrocytes appears outmoded in light of exciting new evidence that astrocytes actively contribute to synaptic plasticity (20–23).
Astrocytes derive from the neuroectoderm and have a complex and heterogeneous morphology, generally described as stellate (i.e., star-shaped; see Figure 2). Each astrocyte occupies its own anatomical domain, in a non-overlapping fashion (24, 25), and has the capacity to interact with the microvasculature and thousands of surrounding synapses (24) (Figure 3). Based on histology, astrocytes can be divided into two major groups: the protoplasmic type in gray matter and the fibrous type in white matter (26). Protoplasmic astrocytes possess a larger quantity of organelles, exhibit short and highly branched processes that envelop synapses, and have “end feet” that cover blood vessels. Fibrous astrocytes have relatively few organelles, exhibit long, unbranched processes that envelop nodes of Ranvier, and physically connect astrocytes to capillary walls. Protoplasmic astrocytes may thus be positioned to influence synaptic transmission, whereas fibrous astrocytes my regulate spike propagation. Further experimental characterization and subclassification of astrocytes is likely to emerge (see below).
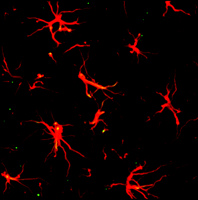
Image of astrocytes in the dorsal horn of a naïve rat spinal cord. Immunostaining of spinal cord sections to reveal astrocytes is accomplished through the use of GFAP antibody and a red fluorescent secondary antibody.
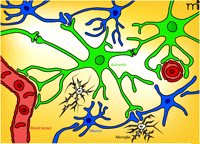
Topographical interrelationships among astrocytes, microglia, and neurons in the CNS. Astrocytes (green) interact with a number of elements: i) capillaries (red; the typical astrocyte endfoot–vessel interaction is indicated); ii) neurons and synapses (blue), thereby giving rise to tripartite synapses; and iii) other astrocytes via gap junctions that permit calcium waves to spread over long distances in the astrocyte network. Astrocytes may release a number of “gliotransmitters” at the tripartite synapses or directly into the extracellular space. Increasing evidence indicates that microglial cells (white) may also be a part of the functional unit of the synapse (thereby constituting a tetrapartite synapse as indicated at bottom of figure). Glial cells are depicted in their resting state. (Oligodendrocytes, which constitute the third major class of glia, are not shown).
Immunohistochemistry with antibodies that bind glial fibrillary acidic protein (GFAP), an astrocyte-specific cytoskeletal protein, is widely used to visualize astrocytes (26) (see Figure 2). The glutamate transporter GLT-1 is another astrocyte-specific marker used to identify astrocytes in the CNS (27, 28). Although S100 calcium binding protein B (S100B) is also used as an astrocyte marker (29), it should be noted that this protein is also expressed by oligodendrocyte progenitor cells that co-express the proteoglycan NG2 (29–31). In the hippocampus, one subpopulation of astrocytes abundantly expresses GFAP, voltage-independent K+ channels, and GLT-1, and is coupled by gap junctions. Another subpopulation expresses voltage-dependent K+ and Na+ channels, S100B, with lower expression of GFAP and GLT-1, and is not coupled through gap junctions (32). In addition, astrocytes from various regions of the CNS (e.g., cerebral cortex or spinal cord) respond distinctively to various substances and neurotransmitters. One recent study also reported a remarkable diversity of astrocyte subtypes in the spinal cord that were identifiable according to a homeodomain code (33). We thus have much to learn about astrocytes and their roles in pain transmission.
Although important physiological functions are attributed to astrocytes in their basal state (23), the differential effects of pathological conditions upon astrocyte function continue to be investigated. Garrison and co-workers (11) suggested the involvement of astrocytes in modulating pain perception, based on an observed correlation between astrocyte hypertrophy in the spinal cord and behavioral pain hypersensitivity following peripheral nerve injury. Astrocyte hypertrophy, whereby a relatively small cell body with thin processes transforms into an enlarged cell body with thickened processes, is generally considered an indicator of activation after injury (Figure 4). In addition to morphological changes, activation of intracellular signaling pathways and release of gliotransmitters (i.e., chemicals released from glial cells that exert their action on neurons or other glia cells) have been attributed to activated astrocytes. Such alterations are currently being explored in the context of enhanced neuronal plasticity and hypersensitivity in various models of pain.
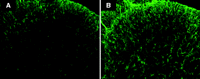
Immunohistochemistry images depicting GFAP immunoreactivity. A) Tissue from the naïve mouse dorsal horn is compared to B) tissue from an animal with collagen-antibody–induced arthritis. Note the increased GFAP signal intensity and astrocyte hypertorphy in the spinal dorsal horn of the animal with arthritis.
Astrocytes in Pain
A number of reports demonstrate activation of astrocytes in the superficial and deep layers of the dorsal horn in models of pain and generally indicate that astrocytes contribute to hypersensitivity. Astrocytes appear to affect spinal processing of nociceptive signaling induced by peripheral inflammation (34), nerve injury (35–39), surgery (40), cancer (41, 42), chemotherapy (43), and spinal cord injury (44, 45). As activation of both microglia and astrocytes is observed, it can be difficult to differentiate the relative contributions of the two cell types to hypersensitivity. It is quite likely that when microglia and astrocytes are concurrently activated, neuronal as well as glial activity is engaged.
There are two sets of circumstances, however, that support a singular role of astrocytes in modulating components of pain transduction. First, it has been shown that the time course of activation of astrocytes after injury differs from that of microglia. In models of neuropathic, inflammatory, and post-operative pain, an increase in the number of activated microglia is observed within a few days, and astrocyte activation becomes visible thereafter; as microglial activation begins to decrease, the astrocyte activation as well as hypersensitivity persist (40, 46–51). In this way, chronic pain may be maintained, rather than initiated, by astrocyte activation, which has important implications for the development of therapeutics. Second, astrocytes show morphological signs of activation that correlate to hypersensitivity in certain pain models in which microglia activity does not appear to change from the baseline state (35, 41, 50). It is important to note, however, that there also are pain models in which microglial activity is elevated without concurrent or subsequent changes in astrocyte activity, indicating that the roles of astrocytes and microglia in modulation of neuronal plasticity are complex and may be variable.
Two drugs (“glia blockers”) that affect both microglia and astrocytes are propentofylline, which is a methylxanthine derivative that attenuates signs of glia activation by an unknown mechanism (52), and fluorocitrate, a glia-specific metabolic inhibitor (53). Intrathecal injection of these drugs into rodents attenuates nociceptive behaviors associated with nerve injury (49, 54, 55), chemotherapy (43), inflammation (49, 56–58), and spinal cord injury (44). Although studies using propentofylline or fluorocitrate do not reveal the differential contribution of microglia versus astrocytes, they do demonstrate that activation of glia can be a necessary component of hypernociception. The intrathecal use of the astrocyte toxin L-alpha-aminoadipate (L-AA), which has no effect on microglial activity, further supports an active role for astrocytes in nerve injury–induced astrocyte hypertrophy and associated allodynia (38, 39).
Although the intermediate filament protein GFAP is used as an astrocyte marker in the CNS, the question remains whether this protein has any functional significance in regulating pain transmission. To address this issue, Kim and coworkers reduced expression of GFAP in the spinal cord of rats by intrathecal injection of GFAP antisense oligonucleotides and also developed GFAP knockout mice (59). They observed that mice lacking GFAP had a shorter duration of tactile allodynia after nerve ligation and that knockdown of GFAP in the spinal cord and dorsal root ganglia (where GFAP is expressed in satellite cells) mitigated nociceptive behaviors in a model of neuropathic pain (59). Because GFAP is important in modulating astrocyte motility (60) and may have important functions in synaptic modulation in the CNS (61–63), it is possible that injury-induced GFAP upregulation not only serves as a marker for astrocyte hypertrophy but that it also contributes to the maintenance of pain states.
There is abundant evidence that various mitogen-activated protein kinases (MAPKs) [e.g., p38, Jun N-terminal kinase (JNK) and extracellular signal regulated kinases (ERK)] play a critical role in the regulation of pain sensitization after tissue and nerve injury (64). In astrocytes, activation of JNK and subsequent downstream signaling appears critically important in regulating pain processing. JNK1and JNK2, but not JNK3, are expressed in the spinal cord, and JNK1 is expressed in astrocytes and neurons (39). Following peripheral nerve injury, however, JNK 1 is activated exclusively in astrocytes (39, 65), and inhibition of spinal JNK by intrathecal injection of the JNK inhibitors SP600125 or D-JNKI-1 (66) attenuates hypersensitivity induced by nerve injury (39, 67, 68). Inhibition of JNK activity in the spinal cord also attenuates hypersensitivity in streptozotocin-induced diabetes. In this model, however, JNK is activated in both neurons and microglia (69).
JNK activates a number of downstream transcription factors, including c-Jun, (which has been detected in astrocytes following peripheral nerve injury), ATF2, c-Myc, and members of the STAT family. Although the critical downstream JNK-induced products that modulate chronic pain remain unknown, the increased expression of monocyte chemotactic protein-1 [MCP-1; also referred to as chemokine-like ligand 2, (CCL2)] has been suggested as one important effector molecule (67). However, neuronal release of spinal MCP-1 following peripheral nerve injury has also been reported (51, 70, 71). Hence, while it appears to play an important role in the regulation of spinal pain transmission, the predominant cellular source of MCP-1 remains unclear.
Activation of Astrocytes by Neurons in Pain Pathways
One of the critical questions concerning the involvement of astrocytes in modulation of nociception is how these glial cells in the dorsal horn sense peripheral injury or inflammation. Given the enhanced activity of small-diameter sensory neurons during injury, it is reasonable to suspect not only that they signal to dorsal horn neurons, but also that they directly or indirectly communicate with astrocytes (Figure 5). Indeed, recent data indicate that astrocyte activation in the dorsal horn during inflammation and following nerve injury is dependent on the inflow of impulses through sensory neurons. The activation of astrocytes (e.g., upregulation of GFAP and IL-1β) in the spinal trigeminal nucleus upon peripheral inflammation is prevented by local anesthetic block of sensory nerve fibers (49). Similarly, early nerve blockade with tetrodotoxin (TTX), a sodium channel blocker obtained from the puffer fish, reduces the expression of GFAP in astrocytes in the cuneate nucleus after median nerve injury (72).
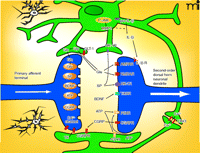
Astrocyte-mediated regulation of synaptic neurotransmission. A number of avenues have been identified whereby astrocytes can influence nociceptive signaling. Some possible targets of astrocyte-mediated control are indicated. See text for details. (AMPAR, α-amino-5-hydroxy-3methyl-4-isoxazole receptor; ATP, adenosine triphosphate; BDNF, brain derived neurotrophic factor; CGRP, calcitonin gene related peptide; CX43, connexin 43; GLT-1, glutamate transporter 1; IL-1β, interleukin 1β; MMP2, matrix metalloproteinase 2; NK-1, neurokinin-1; NMDA, N-methyl-D-aspartic acid; P-JNK, phospho-Jun N-terminal kinase; P2Y, purinergic receptor 2Y; SP, substance P; TrkBR, BDNF receptor; TS, thrombospondin.)
Nociceptive sensory neurons synthesize and release a number of transmitters and neuromodulators, the most well established being glutamate, ATP, SP, and CGRP. Increasing evidence suggests that these substances, as well as NO, galanin, and others, contribute to astrocyte activation (see below). The relative importance of each of these transmitters and the various receptors activated by them have not been determined. Furthermore, as both astrocytes and neurons can release and respond to some of the same substances, (e.g., glutamate and ATP), it is difficult to determine the importance of astrocytes alone in the transmission process.
Current understanding about which transmitters mediate astrocyte activation in the spinal cord is based in part on examination of the cerebral cortex or other brain regions. As discussed above, however, astrocytes from different CNS regions may differ markedly in receptor expression and other properties (73–76). For example, cultured astrocytes from human spinal cords have been reported to have six times more neurokinin 1 receptors per cell than astrocytes from the brain (76). Furthermore, results with astrocytes grown in culture could be dependent on culture conditions as well as the origin of the cells. Despite these potential limitations in interpreting a number of studies, each of the above mentioned transmitters that are released from nociceptive sensory neurons can, independently or in concert, activate astrocytes (e.g., elicit changes in morphology, GFAP expression, and proliferation) in cell culture and spinal cord slice experiments (77–79). Similarly, glutamate can stimulate the release ATP, and vice versa (80, 81), which may be a mechanism for amplification of astrocyte activation in the dorsal spinal cord.
Glutamate
Glutamate release from the terminals of small-diameter sensory neurons with activation of AMPA receptors on dorsal horn neurons is likely the major means of communicating acute noxious information from the periphery to the spinal cord. NMDA receptors, on the other hand, play a pivotal role in facilitating excitation of dorsal horn neurons in response to sustained noxious input (6). Different kinds of glutamate receptors also are present on astrocytes that envelop synapses in the dorsal horn and generate inward currents in response to glutamate, NMDA and kainate (82). Glutamate induces ATP release from cultured brain astrocytes (83, 84), and studies of rat spinal cord astrocytes in vitro indicate that the glutamate receptors involved in generating glutamate-evoked inward currents and ATP-release are primarily of the AMPA class (81). When SP is co-administered with glutamate, the effects are mediated by activation of NMDA and metabotropic glutamate receptors, and the release of ATP is greatly potentiated (81).
Other studies of rat spinal cord astrocytes indicate that glutamate stimulates calcium release from internal stores via an activation of the quisqualate subtype of glutamate receptors (85). There is thus ample evidence that glutamate, acting on different classes of receptors on astrocytes, is of major importance for the interaction between neurons and astrocytes in the dorsal horn.
Adenosine Triphosphate
ATP is a transmitter or cotransmitter in the majority of neurons in the central and peripheral nervous system, and purinergic receptors, represented by several families, are arguably the most abundant receptors in living organisms (86). Astrocytes express both ionophoric P2X and metabotropic P2Y receptors (86). In rat brain astrocytes grown in culture, ATP and other purinoceptor agonists induce morphological changes and increase expression of GFAP and immediate early genes such as c-fos, c-jun and Tis11 (78, 87). ATP (and glutamate) released from neurons may induce calcium oscillations in astrocytes (88–90), and ATP, released from astrocytes themselves, is a very important extracellular mediator for propagation of calcium waves in astrocyte networks (91). ATP enables calcium waves to propagate even when the distance between adjacent astrocytes precludes functional gap junctions (92). In a study of rat dorsal spinal astrocytes in culture, the distinct P2Y1 and P2Y2 subtypes of metabotropic purinoceptors mediate slow and fast responses to ATP, respectively, and are necessary for propagation of calcium waves (93).
Substance P
Substance P is the prototypical neuropeptide, synthesized and released from a subpopulation of nociceptive sensory neurons, and recent studies illustrate the importance of SP in neuronal–glial communication in the dorsal horn. SP receptors (i.e., the neurokinin 1 receptor) are known to be present on cortical astrocytes (94, 95), and over twenty years ago, Mantyh and collaborators reported the specific upregulation of SP receptor binding one to three months after transection of the optic nerve in rabbits. Based on cell morphology, the group concluded that the increase in SP binding was localized to astrocytes (96). The relative abundance of neurokinin 1 receptors on spinal cord astrocytes, as opposed to astrocytes harvested from the brain (76), supports the finding that SP induces phospholipid turnover and increases prostaglandin release in astrocytes from the spinal cord but not in those derived from the cortex or cerebellum (74, 97).
Substance P activates astrocytes cultured from the spinal trigeminal nucleus, as manifested by the upregulation of GFAP and IL-1β (49). This effect is blocked by inhibition of nitric oxide syn-thase (49), which is consistent with the notion that NO regulates the expression of GFAP in mouse primary astrocytes (98) and that the activating effect of SP on astrocytes involves NO (99). Substance P also has been reported to potentiate the IL-1β–induced release of IL-6 and PGE2 from human spinal astrocytes in culture without affecting the IL-1β-induced release of other cytokines. Furthermore, SP enhances the TNF-induced release of IL-6 (76). In this way, SP contributes to the activation of astrocytes, influencing subsequent neuronal excitability and augmenting nociception.
Calcitonin Gene–Related Peptide
CGRP is a well-established peptide neuromodulator in nociceptive sensory neurons (100) and, like SP, contributes to astrocyte activation in the dorsal horn as well as in other brain regions (77, 101). CGRP potentiates the effects of ATP on rat brain astrocytes in primary culture (87) and on its own directly activates signaling pathways in astrocytes (77, 101). For example, CGRP receptor activation elicits the ERK signaling cascade, which in turn increases expression of various downstream signals, such as the cytokines IL-β and TNF, and could increase the excitability of sensory neurons and dorsal horn neurons that participate in spinal sensitization.
Chemokines
Chemokines are low molecular–weight cytokines that coordinate inflammatory responses and promote the pathogenesis of chronic pain (102). Some chemokines are liberated from sensory nerve terminals in the dorsal spinal cord in response to nerve trauma and inflammation (103). The chemokine MCP-1 is upregulated in sensory neurons and transported to their terminals in the dorsal horn following peripheral nerve injury (51, 71, 104). Mice lacking chemotactic cytokine receptor 2 (CCR2), the major receptor for MCP-1 (105, 106), exhibit less activation of microglia and astrocytes and fewer nociceptive responses following nerve injury (106). As discussed above, MCP-1 is also produced by astrocytes after peripheral nerve injury, and CCR2 has been identified on both microglia and neurons (67). Hence, MCP-1 may serve as a neuron-to-microglia, astrocyte-to-neuron, or astrocyte-to-microglia messenger.
Inhibition of CX3CR1, which is the receptor for the chemokine CX3CL1 (i.e., fractalkine), reduces arthritis-induced activation of astrocytes in the rat dorsal horn, suggesting a role for CX3CL1 in astrocyte activation, although this effect could also be mediated indirectly via fractalkine actions on microglia (107–109). Following nerve injury (ligation of the spinal L5 nerve), CX3CL1 expression is induced de novo in astrocytes and upregulated in microglia in the dorsal horn (110, 111). These data indicate that chemokines contribute to activation of astrocytes and possibly to the interaction between astrocytes and microglia in certain forms of neuropathic and inflammatory pain. Clearly, these substances, along with neuromodulatory roles of the other molecules discussed above, contribute to the fine tuning of communication that occurs between the periphery and glia in the spinal cord. Growing evidence indicates that astrocytes in particular release chemicals (see below) that change the environment of the spinal cord and thus contribute to spinal sensitization.
Astrocyte-Derived Factors in Neuronal Activity and Hyperalgesia
Many factors that are released by astrocytes in the dorsal horn and implicated in the regulation of pain pathways are indicated in Figure 5. These substances can excite and/or sensitize presynaptic endings of primary sensory neurons or dorsal horn neurons through a variety of mechanisms, including posttranslational modification, and the upregulation of neuronal receptors, ion channels, and enzymes, ultimately promoting long-term changes in neuronal excitability. The factors and mechanisms by which activated astrocytes enhance neuronal transmission of nociceptive information are far from fully understood. Here, we describe some of the potential substances and mechanisms proposed to mediate the ability of astrocytes to modulate pain perception.
Cytokines
Proinflammatory agents in response to various noxious stimuli include TNF, IL-1β, IL-6, transforming growth factors, and pros-taglandins (22, 112). Several of these factors have been shown to contribute to spinal sensitization in animal models of inflammatory and neuropathic pain (113–122). Peripheral nerve injury, inflammatory insult, and bone cancer can lead to a prolonged increase in IL-1β expression in the dorsal horn (34, 113), and this expression appears to be restricted to astrocytes (49, 123, 124), although some reports indicate that IL-1β may also be released from spinal microglia (125, 126). But given that IL-1β receptors are expressed on dorsal horn neurons (116), IL-1β released from astrocytes might directly modulate neuronal activity. Accordingly, electrophysiological studies show that IL-1β facilitates NMDA receptor activation in dorsal horn neurons (49, 127) and enhances synaptic transmission and neuronal activity in lamina II neurons in spinal slice preparations (116). The notion that spinal IL-1β plays a critical role in hypersensitivity is supported by experiments showing that intrathecal administration of IL-1β produces significant hyperalgesia and mechanical allodynia (115, 120, 128). Furthermore, blocking the action of spinal IL-1β through intrathecal injection of IL-1 receptor antagonists or an IL-1β-neutralizing antibody attenuates hypersensitivity (46, 49, 124, 129, 130). The relevance of IL-1β is a potential mediator of astrocyte-to-neuron communication continues to be investigated.
S100B
Although S100B is also synthesized and released by oligodendrocytes, and radial glial cells, multiple studies suggests that astrocytes are the main source of this calcium binding protein in the CNS (131, 132). S100B may have multiple functions, including regulation of cellular energy metabolism, cytoskeleton modification, and cell proliferation and differentiation. Of particular relevance to nociception, S100B drives the expression cyclooxygenase-2 (COX-2), thereby promoting the generation of PGs, which are thought to play an important role in spinal sensitization (133). S100B binds to and activates the receptor for advanced glycation end (RAGE) products (134) that have been implicated in signaling cascades involved in neuronal synaptic plasticity (135). Interestingly, S100B mRNA and protein are elevated in the spinal cord following peripheral inflammation and nerve injury (123, 136). S100B knockout mice display decreased mechanical allodynia, whereas mice that overexpress the S100B display increased mechanical allodynia in response to peripheral nerve injury (136).
Reduction in GLT-1 Expression
The rapid clearance of glutamate from the synaptic cleft is essential for signal termination, neurotransmitter recycling, and the prevention of excitotoxicity. To date, five glutamate transporters (GLTs) have been identified and characterized. The glial glutamate transporters GLT-1 and glutamate–aspartate transporter (GLAST) (also called excitatory amino acid transporter 2 and 1, respectively) are the major glutamate transporters in the CNS, and GLT-1 expression is largely restricted to astrocytes (137). In the forebrain, GLT-1 is responsible for as much as 90% of the total glutamate transport. Consequently, astrocytes may play a critical role in limiting extracellular glutamate concentrations, and reductions in astrocyte GLT-1 expression in the dorsal spinal cord could accordingly contribute to spinal sensitization and hyperalgesia. Significantly, astrocyte activation in the dorsal horn upon nerve injury is associated with decreased GLT-1 expression (138–142). A similar downregulation of GLT-1 expression is seen in rats with paclitaxel-induced hyperalgesia (143). Propentofylline, a general glia inhibitor, has an antinociceptive effect in nerve injury that is correlated with its ability to restore GLT-1 expression (52, 55). Recombinant adenovirus-mediated gene transfer of GLT-1 into the spinal cord, forcing astrocytes to express GLT-1, reduces both inflammatory and neuropathic pain (144). Therapeutic interventions to promote GLT-1 activity might thus be useful for the treatment of neuropathic pain.
Glutamate
Upon its uptake by astrocytes, glutamate is metabolized to glutamine by glutamine synthetase. Newly synthesized glutamine is shuttled back into neurons, where it can be reconverted to glutamate (145). It has further been suggested that the calcium-dependent release of glutamate from astrocytes affects neuronal hyperexcitability via extrasynaptic NMDA receptors (146, 147). Further studies are warranted to appraise the direct contribution of glia-derived excitatory amino acids upon central sensitization and hyperalgesia.
Thrombospondin
Astrocyte processes are in close contact with neuronal cell bodies and enwrapped active synaptic terminals and astrocytes promote synapse formation and maturation (148–150). The astrocyte processes display a high degree of motility and dynamic morphological changes, which may be important for correct placement of gliotransmitter release and proper positioning of processes containing glutamate transporters. Time-lapse studies have shown that astrocytes can rapidly extend and retract fine processes to engage and disengage from motile postsynaptic dendritic spines (151), indicating that the astrocyte ensheathment and modulation of synaptic function is a dynamic, rather than static, process. Thrombospondin, ephrin A3, and ephrin A4 released from astrocytes have been implicated in synaptogenesis in a temporal and regional-specific manner (149, 152). Although the role of thrombospondin in synaptogenesis has been appreciated for some time, only recently has it been demonstrated that thrombospondins bind to the α2δ-1 subunit of voltage-gated calcium channels (153). Both in vitro and in vivo experiments show that the drug gabapentin antagonizes the binding of thrombospondin to α2δ-1 and inhibits excitatory synapse formation (154). The role of α2δ-1 in excitatory synapse formation and the therapeutic function of gabapentin in blocking astrocyte-mediated synapse formation are exciting areas of research.
Astrocytes as Targets in Clinical Pain Control?
Targeting astrocyte function as a novel strategy to control chronic pain states is an evolving concept. Therapeutic approaches to block astrocyte communication might include: i) a general disruption of astrocyte function; ii) inhibition of intracellular signals that drive the synthesis of pronociceptive substances in astrocytes; iii) neutralization of factors that are released from activated astrocytes; and iv) antagonism of astrocyte-derived nociceptive substances at neuronal receptors. Preclinical studies using pharmacological tools to disrupt astrocyte function and attenuate nociception in animal models of pain are promising. Most of these drugs, however, are not appropriate for human application, and so clinical assessment of astrocyte inhibition has not been achieved. Clinical efforts to develop a therapeutic “astrocyte-inhibitory” strategy directed at controlling pain will have to be measured against the important homeostatic functions effected by astrocytes. For example, intrathecal injection of fluorocitrate, an inhibitor of astrocyte and microglial cells, attenuates hypersensitivity in animal models of pain, but the drug functions by blocking glial uptake of excitatory amino acids, an effect that has obvious implications in terms of neuroexcitability, seizures, and neurotoxicity.
Given the dorsal horn astrocyte-dependent increase in IL-1β expression in various animal models of pain (34, 49, 113, 123, 124), spinal IL-1β represents a potential target for treating chronic pain syndromes. Blockade of spinal IL-1β actions has been shown to be antinociceptive in animal models of pain, and a clinically approved IL-1β antagonist (anakinra) is available. Similar effects have been observed after spinal delivery of the clinically approved TNF-decoy receptor etanercept (117, 155). With the increasing number of available cytokine blockers, it will be interesting to learn whether those targeting astrocyte-associated factors have pain-relieving effects. However, given that the biological modifiers that are currently available do not readily cross the blood–brain barrier (BBB), the assessment of such effects in humans remains a challenge.
Propentofylline, pentoxifylline, and ibudilast are methylxanthine derivatives that act as phosphodiesterase inhibitors. These drugs suppress astrocyte activation and attenuate pain behavior in animal models of various pain states, but the mechanisms accounting for these actions are yet to be determined. What is intriguing is that these drugs reduce spinal glia activation when given systemically (43), and they are already in clinical use for other indications. Ongoing clinical studies will reveal if these drugs will prove useful in the treatment of chronic pain.
In addition to current studies that are focused on blocking the action of astrocytes and microglia, it should be noted that activation of glia in the CNS may also subserve therapeutic strategies. Glial activators could possibly act to regulate neuronal excitability and restore normal pain signaling (156, 157). Thus, in order to provide new glia-associated avenues for the development of pain therapeutics, it will be critical that we fully investigate the protective, normalizing, and physiological actions of astrocytes as well as the pathological ramifications of these intriguing CNS cells.
Conclusions
Chronic pain is a major health problem, affecting approximately twenty percent of the population and markedly reducing the quality of life for patients and their relatives. At several additional levels of society, chronic pain extols extensive costs. Drug development in the area of chronic pain thus addresses a crucial need, as there are currently few available effective treatments for most chronic pain conditions. In the past, drugs to treat pain conditions have been developed to target neurons, a logical extension of our understanding of chronic pain signaling as the disruption of communication between the site of injury and the CNS. However, glial cells have recently become recognized as important modulators of neuronal activity that contribute to the induction and maintenance of pathological pain states. There are an increasing number of studies showing that astrocytes express receptors for multiple factors released from neurons in response to peripheral painful insults and that astrocytes are dynamically responsive to their microenvironment. The activation of astrocytes is reflected in specific changes in gene expression and signaling via neurotrophic, neuromodulatory, and proinflammatory mediators. Factors such as glutamate, thrombospondin, and IL-1β which augment synaptic activity in the dorsal spinal cord, have been linked to astrocyte function and chronic pain. A role for astrocytes in pain modulation is also supported by pharmacological studies in animals. Further investigation of astrocytes will likely have great bearing on the development of experimental therapeutics for pain management and for our understanding of neuroregulatory mechanisms in pain and disease.
Acknowledgement
We thank Prof. Michael R. Vasko for critical reading and editing the manuscript, and Drs. Simone Codeluppi and Johanna Petterson for providing images of spinal cord astrocytes. This work was supported by the Swedish Research Council (CIS), Marie Curie International Reintegration grant (CIS) and International Association for Studies of Pain (CIS).
- Copyright © 2010
References

Ernst Brodin, PhD, has pursued pain-related preclinical research at the Karolinska Institute since the early 1980s. He became professor in Pharmacology in 2000. His research has dealt mostly with the role of substance P and other neuropeptides in pain mechanisms. He has recently begun to focus on the possible role of supraspinal glial activation in mechanisms of chronic pain.

Camilla I. Svensson, PhD, is an assistant professor at the Department of Physiologoy and Pharmacology at the Karolinska Institute in Sweden. Her research is focused on pain mechanisms in conditions of chronic inflammation, and she has a special interest in the role of spinal glia in pain transmission.

