Have Tumor Suppressor PDCD4 and its Counteragent Oncogenic miR-21 Gone Rogue?
MicroRNAs (miRs) are non-coding RNAs that control gene expression by binding to specific sites on the 3′ UTR of target mRNAs and causing translational repression. miR-21 has emerged from many profiling experiments as a miRNA whose expression is dys-regulated in cancer (1). Increased miR-21 expression is observed during carcinogenesis, in tumors, and in response to chemotherapy (2). Causal links between miR-21 expression and cellular proliferation, migration, apoptosis, tumor growth, invasion and metastasis have been established (3, 4), and increased miR-21 activity has been detected in brain, breast, colon, ovarian, lung, pancreatic, and other cancers (1–3). Furthermore, miR-21 activity appears to be increased in many inflammation-induced conditions, such as lipopolysaccharide (LPS)-induced mouse lung inflammation, allergic airway inflammation, osteoarthritis, psoriasis, ulcerative colitis, cardiac muscle injury, and cardiac hypertrophy (5). Components of inflammation are known to include pro-inflammatory cytokines and reactive oxygen species, both of which appear to drive tumor progression as well as other conditions (6). Therefore, as speculated by Sheedy et al. (7), miR-21 may be a link between cancer and inflammation.
Programmed Cell Death 4 (PDCD4) is a well-documented tumor suppressor that inhibits neoplastic transformation, tumor progression, and translation (8, 9). PDCD4 interacts with factors involved in translation initiation, such as RNA helicase eIF4A and scaffold protein eIF4G, and inhibits the initiation of protein translation in an mRNA-selective fashion (10–12). In addition to its role in translation, PDCD4 can shuttle between the cytoplasm and the nucleus (13, 14). The expression of PDCD4 is increased during apoptosis (8, 9) and decreased during human and mouse carcinogenesis (15–17). Overexpression of PDCD4 inhibits tumorigenesis and tumor progression in a transgenic-mouse model and inhibits tumor cell invasion in vitro (8, 9, 16–18). PDCD4-null mice show spontaneous development of lymphomas (19) as well as enhanced sensitivity to carcinogen-induced skin cancer (17). PDCD4 has emerged as a major, functionally significant target of miR-21 (9, 20, 21). The observed inverse correlation between PDCD4 and miR-21 expression may be a useful prognostic marker (9, 14). PDCD4 and miR-21 are potential targets for novel cancer prevention or anti-cancer therapies (8, 9).
The recent study by Sheedy et al (7) makes the observation that, unlike wild-type mice, PDCD4-null mice are resistant to the killing effects of the Toll-like Receptor 4 (TLR4) ligand LPS, a bacterial endotoxin. In probing the responsible pathway both upstream and downstream of PDCD4 (Figure 1), the authors provide evidence supporting a pathway through TLR adaptor protein MyD88 and transcription factor NF-κB, leading to increased miR-21 expression and decreased expression of PDCD4. The loss of PDCD4 in this model results in increased expression of interleukin (IL)-10 and decreased NF-κB activity (see Figure 1). Elevated amounts of IL-10 and IL-4 were previously observed by one of the authors in describing PDCD4-null mice (19). The results presented by Sheedy et al. establish that: 1) PDCD4 is an important mediator of the killing response to LPS (survival is 30% for wild type vs 70% for PDCD4 null mice); 2) LPS induces miR-21 expression by a process that requires MyD88 and the NF-κB subunit p65, as bone marrow–derived macrophages (BMDMs) deficient in MyD88 and mouse embryonic fibroblasts lacking p65 failed to induce miR-21 in response to LPS; 3) PDCD4 protein expression is lost in response to LPS by a process that involves both miR21-mediated inhibition of PDCD4 translation, and proteasome-dependent protein degradation that is mediated (as shown previously by others) by Akt–mTOR signaling (17, 22); 4) consistent with a previous report of Yang et al. (15), PDCD4 deficiency leads to inhibition of NF-κB dependent transcription in LPS-treated HEK293 cells and a decrease in NF-κB regulated IL-6 expression in knockout mice and PDCD4-deficient BMDMs; and 5) PDCD4 deficiency in mice and BMDMs derived from these mice leads to an increase in IL-10 protein but a decrease in IL-10 mRNA (7). The authors suggest that this observation is consistent with IL-10 being a translational target of PDCD4.
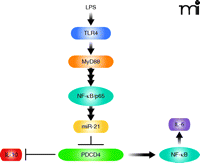
TLR4 signaling: The MyD88-dependent pathway through PDCD4. LPS activates TLR4 and stimulates activation of the MyD88-dependent early-phase activation of NF-κB, which leads to an increase in miR-21 expression. miR-21 inhibits PDCD4 expression via translational regulation. PDCD4 regulates IL-10 expression by post-transcriptional regulation, possibly by inhibiting translation. PDCD4 appears to positively regulate NF-κB activity and consequent IL-6 expression.
If validated, IL-10 may be one of a relatively few translational targets of the inhibitor of translation initiation PDCD4. Carbonic anhydrase type II may also be translationally regulated in HEK293 and in Bon-1 cells overexpressing PDCD4 (23). Several pretranslational targets are functionally significant in the mechanism by which PDCD4 suppresses tumorigenesis or invasion, among them the matrix modulators urokinase-type plasminogen activator receptor (uPAR), tissue inhibitor of metalloproteinase-2 (TIMP-2), and lysyl oxidase, as well as the Jun N-terminal kinase (JNK) activator MAP4K (8, 9, 18, 24), (Figure 2). Because PDCD4 interacts directly with translation initiation factors eIF4A and eIF4G, the direct targets of PDCD4 are expected to be translational. Sheedy et al.’s observation that PDCD4 deficiency increased IL-10 protein but not RNA levels is consistent with a posttranscriptional, possibly translational target (7). The significance of the oppositely regulated IL-10 mRNA levels is not clear. Additional experiments would be called for to exclude posttranslational loss of IL-10. Positive evidence for IL-10 as a translational target might include identification of a shift of IL-10 mRNA out of actively translating polysomes with increasing amounts of PDCD4 present. Cotransfection of a reporter containing the IL-10 UTRs (but not IL-10 promoter) with increasing amounts of PDCD4-expressing vector would also be useful for determining whether IL-10 is translationally regulated.
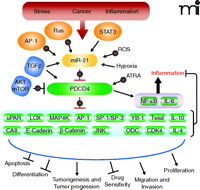
Therapeutic Targeting of miR-21 and PDCD4: Anti-Inflammatory vs Anti-Cancer. Deregulated miR-21activity occurs under many conditions, including stress, cancer, and inflammation. Although miR-21 expression is regulated by transcription factors such as AP-1 and STAT3, most miR-21 regulation takes place post-transcriptionally. miR-21 RNA expression is activated by TGFβ, reactive oxygen species (ROS), Ras, all-trans retinoic acid (ATRA), and hypoxia. PDCD4 translation is inhibited by miR-21 and PDCD4 is post-translationally regulated by AKT–mTOR–dependent proteasomal degradation. Direct (translationally regulated) targets of PDCD4, including carbonic anhydrase II (CAII) and IL-10, still need to be validated. Indirect targets (targets that appear to be pre-translationally regulated by PDCD4) include: urokinase receptor (uPAR), lysyl oxidase (LOX), mitogen-activated protein kinase kinase kinase kinase 1 (MAP4K), Twist1, Y-Box-binding protein (YB-1), E-cadherin, β-catenin, ornithine decarboxylase (ODC), cyclin-dependent kinase 4 (CDK4), and Jun N-terminal kinase (JNK). In addition, transcription factors AP-1, SP-1, and SP-3 are negatively regulated by PDCD4, whereas NF-κB is positively regulated by PDCD4. Increased expression of PDCD4 and decreased expression or availability of PDCD4 targets can lead to inhibition of proliferation, migration, invasion, tumorigenesis, and tumor progression. Increased expression of PDCD4 can also increase apoptosis, drug sensitivity, differentiation, NF-κB activity, and inflammation. Bulls-eye targets indicate possibilities for therapeutic intervention.
A previous report showed that IL-10, IL-4, and interferon (IFN)-γ were elevated in PDCD4-deficient splenocytes, as compared to those levels found in wild-type cells, grown in vitro on plates containing CD3- and CD28-specific antibodies (19). Not clear however, is the basal amount of IL-10 in PDCD4-deficient macrophages. Additionally, amounts of IL-6 mRNA and protein were lower in PDCD4-deficient mice and in PDCD4-deficient BMDM after LPS exposure, again with no information about basal amounts. Also not shown are the amounts of other cytokines known to be induced by LPS, such as tumor necrosis factor–α (TNFα), and whether they are operative in LPS-induced killing. IL-10 concentrations in blood are about twice as high in PDCD4-null mice as in wild-type mice at one-hour post LPS injection. IL-10 protein is about 1.7-fold higher in PDCD4-null BMDMs treated with 10 or 100 ng/ml of LPS. Thus, although it is apparent that IL-6 is transcriptionally regulated and IL-10 is post-transcriptionally regulated by PDCD4, it would be important to know whether PDCD4 counteracts LPS induced changes in these cytokines.
Early-phase NF-κB activation by LPS-activated TLR requires MyD88 whereas late-phase NF-κB activation is independent of MyD88 (25). The expression of IL-6 and other cytokines is elevated during the early response, whereas the expression of IFN-β and IFN-inducible genes increases later. From the decrease in IL-6 transcription and the reduced (NF-κB-dependent) resynthesis of IκBα (an inhibitor of NF-κB) after LPS exposure in PDCD4-deficient BMDMs, Sheedy et al. conclude that PDCD4 positively regulates NF-κB activity (7). Furthermore, because this regulation occurs without affecting degradation of IκBα, the authors conclude that this regulation of NF-κB is probably indirect or secondary to PDCD4 inhibition of translation. Interestingly, Sheedy et al. showed that LPS induced miR-21 expression is dependent on NF-κB/p65 (7). IL-6 regulated activation of STAT3 is known to induce miR-21 expression (26); thus, TLR4/MyD88 dependent activation of NF-κB and expression of IL-6 could lead to a STAT3-dependent increase in miR-21. This would create a negative feedback loop in which miR-21–mediated inhibition of PDCD4 translation would lead to decreased NF-κB activity.
The events responsible for LPS-dependent cell killing are not clear but are thought to involve other NF-κB–regulated pro-inflammatory cytokines, such as TNFα and IL-1β (25). Are the expressions of these cytokines also decreased in PDCD4-deficient mice and in their BMDM? Knowing this answer would help to determine whether the LPS-protective effects of PDCD4 gene deletion are linked to the decreased expression of IL-6 and increased expression of IL-10. Further investigation is also needed to determine the significance of these downstream targets of PDCD4 in carcinogenesis. Inflammation is a promoter of carcinogenesis (6), so we must ask whether and how the “pro-inflammatory” actions of tumor suppressor PDCD4, namely increased NF-κB and IL-6 expression and decreased IL-10 expression, might be drivers of carcinogenesis. Clearly, there are differences in LPS signaling through miR-21–PDCD4 and the miR-21–PDCD4 signaling that mediates the process of carcinogenesis.
Is the miR-21/PDCD4 pathway a good target for therapeutic intervention? Sheedy et al. speculate that targeting the miR-21–PDCD4 pathway might provide new therapeutic possibilities for boosting or limiting TLR4 activity, as necessary, for vaccine adjuvancy or for treatment of inflammatory disease (7). Similarly, others have speculated that the miR-21–PDCD4 pathway provides a good target for cancer prevention and intervention (8, 9). Figure 2 shows several possibilities for molecular targeted intervention. Because elevated amounts of miR-21 appear to be functionally significant in a number of inflammation-associated conditions including cancer, targeting the increased expression of miR-21 might be envisioned. The use of agents such as anti-miR-21 nucleotides might be useful. miR-21 transcription is induced by transcription factors STAT-3, AP-1, and NF-κB, as well as by Ras signaling and oxidant stress (1). Agents that target these pathways are in the clinic or in clinical trials for cancer prevention or treatment. To counteract the loss of PDCD4 caused by elevated miR-21, morpholino-21, which specifically protects PDCD4 mRNA, might be considered. For cancer prevention, counteracting the miR-21 effect on PDCD4 might be advantageous, but one should bear in mind the alternative pathway for PDCD4 loss that is driven by elevated PI3 kinase–Akt signaling (27). In summary, the data presented by Sheedy et al. suggest the need to proceed with caution in designing new therapies that target the miR-21–PDCD4 pathway (7). One needs to ensure that anti-cancer interventions that diminish miR-21 or increase PDCD4 do not also induce an adverse inflammatory response. Conversely, steps need to be taken to ensure that therapies designed to regulate TLR4 by increasing miR-21 and decreasing PDCD4 do not also create increased risk factors for carcinogenesis.
- Copyright © 2010
References

Nancy Colburn, PhD, is Chief of the Gene Regulation Section, LCP, and Chief of the Laboratory of Cancer Prevention at the NCI. Her laboratory has been an international leader in the discovery of molecular events that drive carcinogenesis and might be targeted for cancer prevention. Among these molecular events are activation of oncogenic transcription factors AP-1 and NF-κB and inactivation of tumor-suppressing translation factor PDCD4. Dr. Colburn’s recent research has extended to dietary interventions to prevent carcinogenesis in mice and humans and the discovery of predictive bio-markers. E-mail colburna{at}mail.nih.gov; fax 301 846-6907.

Noriko Yoshikawa, PhD, is a postdoctoral fellow in the Gene Regulation Section, LCP. She received her PhD from Mukogawa Women’s University, Japan. Her research is focused on generating tools for discovery of Pdcd4 targets and characterizing these targets. E-mail yoshikawan@mail. nih.gov; fax 301 846-6907.

Arti Santhanam, PhD, is a senior postdoctoral fellow in the Gene Regulation Section, LCP. She received her PhD from the New Jersey College of Medicine and Dentistry. Her research addresses the mechanism, the structure, and the genes targeted by the tumor suppressor PDCD4 when it prevents breast cancer–cell invasion. E-mail santhana{at}mail.nih.gov; fax 301 846-6907.

Matthew Young, PhD, is Staff Scientist in the Gene Regulation Section of the Laboratory of Cancer Prevention, Center for Cancer Research, NCI, NIH. He received his PhD at the University of Maryland and postdoctoral training at the National Cancer Institute. His research focuses on mouse and human studies designed to provide molecular targeted prevention of carcinogenesis and on the discovery of predictive biomarkers. E-mail young-ma{at}mail.nih.gov; fax 301 846-6907.

