Finding Homes for Orphan Cytochrome P450s: CYP4V2 and CYP4F22 in Disease States
Abstract
Genetic analyses have identified a wide spectrum of mutations in the CYP4V2 gene from patients suffering from Bietti’s crystalline corneoretinal dystrophy, and mutations in the CYP4F22 gene have been linked to lamellar ichthyosis. These strong gene–disease associations will be better understood if we can elucidate the substrate specificity of the heretofore “orphan” P450s and unravel the biochemical pathways that go awry in disease states. The complex biotransformations that underlie eicosanoid signaling, however, pose great challenges for the enzymologist seeking to assign specific metabolic roles to these members of the CYP4 family. Inductive reasoning and modeling are crucial tools for designing the experiments that will define disease progression in terms of CYP function.
The cytochrome P450 (CYP) 4 family of enzymes contains several recently identified members that are referred to as “orphan P450s” because their endogenous substrates are unknown. Human CYP4V2 and CYP4F22 are two such orphan P450s that are strongly linked to ocular and skin disease, respectively. Genetic analyses have identified a wide spectrum of mutations in the CYP4V2 gene from patients suffering from Bietti’s crystalline corneoretinal dystrophy, and mutations in the CYP4F22 gene have been linked to lamellar ichthyosis. The strong gene–disease associations provide unique opportunities for elucidating the substrate specificity of these orphan P450s and unraveling the biochemical pathways that may be impacted in patients with CYP4V2 and CYP4F22 functional deficits.
Introduction
Cytochromes P450 (P450s) are a superfamily of heme-containing monooxygenases that catalyze a myriad of oxidative metabolic reactions through generation of high-valent iron-oxo porphyrin radical intermediates (1). Humans possess fifty-seven functional P450s, many of which have well-established roles in the metabolism of endogenous sterols, bile acids, vitamins, and fatty acids, as well as in the metabolic clearance of drugs and other xenobiotics. Thirteen human P450s have been designated as “orphans” because their functional roles have yet to be elucidated (2). Among these thirteen enzymes, almost half—CYP4A22, CYP4F11, CYP4F22, CYP4V2, CYP4X1, and CYP4Z1—belong to the CYP4 family of enzymes. This review focuses on CYP4V2 and CYP4F22 and the prospects for “de-orphanizing” these two enzymes through an understanding of their roles in human ocular and skin disease.
Enzymes of the CYP4 family are traditionally associated with endogenous fatty acid metabolism, as discussed in several recent reviews (3–5). For example, CYP4F3A, present in neutrophils, has an essential role in catalyzing the ω-hydroxylation of leukotriene B4, thereby abrogating the latter’s chemotactic properties. CYP4A11 and CYP4F2 both metabolize arachidonic acid to 20-HETE, and their relatively high expression in the kidney has stimulated investigation into the pharmacogenetics of these enzymes in relation to hypertension, although such a relationship remains controversial. In contrast, genetic polymorphisms in CYP4V2 and CYP4F22 are strongly associated with Bietti’s crystalline dystrophy (6) and lamellar ichthyosis, respectively (7). We speculate here that both of these orphan P450s may have important roles in eicosanoid metabolism and/or signaling processes. Indeed, cytochrome P450 enzymes from all of the CYP1–4 families are well-recognized catalysts of ω-3 and ω-6 oxidations of polyunsaturated fatty acids (PUFAs) (8). Considerable difficulties exist in validating physiologically relevant metabolic roles for orphan P450s (9). However, these two genetically defined disorders provide new opportunities for tackling this problem by providing an expanded array of biologically relevant models (e.g., tissue samples from affected patients) that can be used in lipidomic analyses.
Linkage of CYP4V2 to a Degenerative Ocular Disease
Bietti’s crystalline dystrophy (BCD) (10) is an autosomal recessive disease characterized by the presence of yellow-white crystals and/or complex lipid deposits in the cornea and retina (Figure 1A), atrophy and degeneration of the retinal pigmented epithelium (RPE), and sclerosis of the choroidal capillaries. These symptoms progress from visual field constriction to eventual night blindness. BCD is a progressive disease, but patients as young as sixteen to seventeen years of age have been diagnosed (11). BCD appears to be more common in East Asia, although cases have been described in patients of Lebanese and Mexican origin (12, 13) as well as in European patients presenting with retinitis pigmentosa (14). Whereas BCD is generally considered to be a rare genetic disease, it has been argued that it may be an underdiagnosed condition on the basis of one study in which approximately ten percent of autosomal recessive retinitis pigmentosa cases were indicative of BCD (14). If such a relatively high incidence of BCD were to prove correct, a case could be made for the development of therapeutic modalities, perhaps using gene therapy, as noted by recent successes in this field (15).

A. Fundus photograph of the eye of a Bietti’s patient illustrating typical yellow crystalline deposits and atrophy of the retinal pigmented epithelium. [From (13), with permission.] B. Hyperkeratotic scaling on the leg of a lamellar ichthyosis patient. (Courtesy of Dr. Philip Fleckman, Department of Dermatology, University of Washington, Seattle.)
Toward the Molecular Etiology of BCD
A decade ago, genetic linkage analysis first identified human chromosome 4q35 as the locus harboring the genetic defect(s) in BCD (16). Fine-mapping of this locus implicated CYP4V2 because potentially disruptive exonic and intronic mutations were identified in twenty-three BCD patients (6). Subsequently, many investigators have corroborated a role for the gene (12, 13, 17–22), with more than twenty discrete disease-associated mutations of CYP4V2 having been described (Figure 2A). The most frequent CYP4V2 mutation (IVS-8delTCATACAGGTCATC) results from alternative splicing at the splice-acceptor site for exon 7. Seventeen base-pairs in the 3′ end of intron 6 and on the 5′ end of exon 7 are removed, and as a result, exon 7 is deleted. This polymorphism, as well as the exon 9 and exon 10 deletions and exon 2 insertions, would be expected to cause gross structural changes in any translated protein, likely to result in complete loss of enzyme activity. Similarly, numerous amino acid deletions and substitutions have been predicted to negatively affect CYP4V2 activity based on their conservation across other CYP4 enzymes and some early homology modeling of CYP4V2 (6).
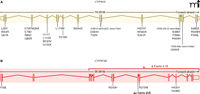
A. BCD-related mutations in CYP4V2 are compiled from ten separate studies; all eleven exons have been found to contain at least one mutation. B. In lamellar ichthyosis (type 3), seven different mutations have been linked to disease (7), with each affected individual homozygous for a single mutation.
We have developed new homology models of human CYP4V2 (Figure 3), using the Robetta server (23, 24) and a 1.2-Å resolution X-ray structure of cytochrome P450 BM3 (PDB ID: 2IJ2) from Bacillus megaterium as a template (25). This analysis suggests that disease-associated amino acid substitutions in CYP4V2 occur throughout the enzyme structure. Two residues (L22, W44) map to the putative N-terminal membrane anchor, several (I111, M123, L173, T325, H331, S341, and R400) are located within the hydrophobic core of the enzyme (toward the heme active center), and the remainder reside at the solvent-accessible surface of the protein (Figure 3). Mutations near the active-site heme center might be expected to diminish intrinsic catalytic activity, whereas those at the exterior of the protein or within the N-terminal anchor domain may be more likely to disrupt protein–protein interactions between CYP4V2 and redox partners (cytochrome P450 reductase and cytochrome b5). Moreover, all structurally important mutations have the potential to reduce catalysis by accelerating enzyme degradation and lowering steady-state concentrations of the protein. However, no experimental data are yet available that address the potential functional consequences of CYP4V2 mutations, and other high-resolution approaches are needed to validate the evolving homology model(s).
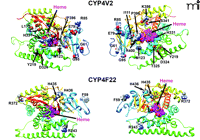
The residues highlighted are those that are sites of BCD-linked mutations of CYP4V2 and lamellar ichthyosis–linked mutations of CYP4F22. (See text for details.)
Substrate Selectivity of Recombinant CYP4V2
CYP4V genes have been identified in humans (CYP4V2), rodents (Cyp4v3), and fish (cyp4v1, cyp4v5–9). CYP4V2 is the most distantly related of the human CYP4 family, possessing only ~35% sequence identity to other family members (5). Despite this relatively low degree of amino acid homology, CYP4V2 expressed in insect cells has been shown to catalyze fatty acid ω-hydroxylation reactions with medium-chain saturated fatty acids at rates that are greatly reduced by HET0016, a CYP4 chemical inhibitor, at nanomolar concentrations (26). Therefore, CYP4V2 displays both the substrate and inhibitor characteristics of prototypical CYP4 enzymes. The catalytic efficiency (Vmax/Km) of CYP4V2-mediated ω-hydroxylation of medium-chain saturated fatty acids in the foregoing study was quite low, but Vmax/Km can vary greatly with the structure of the fatty acid substrate involved. For example, CYP4F3A ω-hydroxylates both arachidonic acid and leukotriene B4, the latter 800-fold more efficiently than the former (27). Therefore, disruption of some key endogenous eicosanoid anabolism or catabolism pathway(s) in BCD seems a reasonable working hypothesis.
Endogenous Fatty Acid Metabolism in BCD
A rational approach to investigating the biochemical basis of BCD and revealing the endogenous substrate specificity of CYP4V2 would be to define the chemical composition of the crystalline deposits that have been observed macroscopically, not only in the eye but also in fibroblasts and lymphocytes derived from BCD patients (27–29). However, the paucity of biological material available for analysis from these deposits, along with the complexity of the lipids that are present, especially in aging tissues, has precluded this approach. Fibroblasts and lymphocytes from BCD patients have been shown to display abnormal lipid metabolism in that synthesis of ω-3 PUFAs; specifically, levels of eicosapentaenoic acid [EPA (i.e., 20:5 n-3)] and docosahexaenoic acid [DHA (i.e., 22:6 n-3)] from α-linolenic acid (i.e., 18:3 n-3) are reduced (30). In contrast, the ω-6 biosynthesis pathway from linoleic acid (i.e., 18:2 n-6) does not appear to be affected. More recently, free fatty acid profiles have been analyzed in BCD patients (31), and these profiles suggest that the synthesis of monounsaturated fatty acids may be impaired, as plasma oleic acid (i.e., 18:1 n-9) levels are significantly reduced in BCD, while palmitic acid (i.e., 16:0) concentrations are concomitantly elevated. Collectively, these studies point to a defect in fatty acid metabolism in BCD, but exactly how these biochemical changes relate to a deficit in CYP4V2 catalysis is far from clear.
ω-3 Polyunsaturated Fatty Acids (PUFAs): Potential Endogenous Substrates of CYP4V2?
No studies have yet examined lipid metabolism in the primary target organ of BCD—the eye, which is notable for having very high concentrations of DHA. Ocular photoreceptor tips, in fact, have the highest DHA content of any cell (32). Photoreceptor cells have a cylindrical shape with outer segments that are embedded in the apical microvilli of RPE cells. Significantly, the outer segments of photoreceptor cells contain membrane disks, rich in rhodopsin as well as phosphatidyl choline and phosphatidyl ethanolamine, both esterified to DHA and other fatty acids (33). The efficient recycling of lipids to regenerate disk membranes is essential to photoreceptor cell function, although the regulatory mechanism of disk membrane recycling is not well understood, especially regarding the processes by which synthesis and disposal of disk membranes are coordinated (34). It is known that about ten percent of the outer segment is renewed daily in primates (35). The cycle begins with shedding of the tips of the outer segments of rods and phagocytosis of the shed material so that lipids from the shed membranes can enter into RPE cells by endocytosis (36). Lipids are then transferred from RPE cells to rod inner segments for the biosynthesis of disk membranes (Figure 4). In addition, DHA formed in the liver from precursor ω-3 fatty acids enters the circulation and is taken up by RPE cells (37). Clearly, RPE cells have an important role in outer membrane-disk regulation and process large amounts of DHA and other fatty acids (38). The mechanisms for regulating PUFA levels in RPE cells thus appear to be highly efficient.
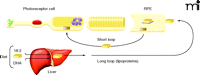
Highly efficient mechanisms for DHA utilization and recycling exist among RPE photoreceptor cells (short loop). These mechanisms complement the more significant uptake of dietary DHA from plasma lipoproteins (long loop). [Adapted with permission from (38).]
In preliminary studies, we have found that CYP4V2 protein is expressed in RPE cells and that recombinant CYP4V2 can hydroxylate the ω-3 PUFAs, DHA, and EPA (M. Nakano, E.J. Kelly, and A.E. Rettie, unpublished results). Interestingly, DHA and EPA are precursors of oxygenated resolvin and protectin metabolites that possess anti-inflammatory and immunoregulatory properties (39). From DHA, the pathways to the D-series metabolites share a lipoxygenase-generated intermediate, 17S-hydroperoxy-DHA, that is reduced to the 16,17-epoxide that is then hydrolyzed to neuroprotectin D1 (Figure 5). A slightly more complex series of reactions yields the trihydroxy metabolite, resolvin D1, also from the 17S-hydroperoxy-DHA intermediate (40). In an analogous fashion, EPA is the progenitor of E series metabolites, such as resolvin E1.

ω-Hydroxylation catalyzed by CYP4V2 may act at one or more levels of either pathway to modulate protectin/resolvin signaling. (See text for details.)
These anabolic pathways have clear similarities to leukotriene B4 formation from arachidonic acid. It is well established that catabolism (inactivation) of leukotriene B4 relies on initial ω-hydroxylation by the neutrophil P450 enzyme CYP4F3A, followed by β-oxidation (41). Therefore, it is possible that similar CYP4-mediated processes could be involved in the inactivation of the resolvins, protectins, and their PUFA precursors. Indeed, the 22-(ω)-hydroxy metabolite of neuroprotectin D1 has been identified in human neutrophils and glial cells exposed to DHA or 17S-hydroperoxy-DHA (42). CYP4 enzymes are, likewise, viable candidates for terminating PUFA-related signaling in the eye, and so it will be important to clarify the expression profile of CYP4V2 and related ω-hydroxylase enzymes in ocular tissues. In summary, we speculate that altered PUFA anabolism/catabolism, secondary to defective CYP4V2 function in RPE cells, may be a contributor to the lesions that develop in BCD.
Linkage of CYP4F22 with a Hyperkeratotic Skin Disease
Ichthyoses are a heterogeneous group of genetic disorders of keratinization characterized by desquamation of the skin (43). The disease has six distinct clinical subtypes: harlequin ichthyosis, lamellar ichthyosis, nonbullous congenital ichthyosiform erythroderma, bullous congenital ichthyosiform erythroderma, X-linked ichthyosis, and ichthyosis vulgaris. Lamellar ichthyosis is characterized by the presence of large, dark, plate-like adherent scales in the absence of erythroderma (Figure 1B), and the estimated incidence is 1 in 300,000 live births. Afflicted newborns are usually encased in a translucent pergament-like collodion membrane, owing to a defective stratum corneum. Several genes are associated with ichthyosis, some of which are responsible for the synthesis of enzymes and transporters involved in the production, transport, or assembly of components of the stratum corneum. Genes that have been associated with the disease include: TGM1, which encodes a transglutaminase involved in the cross linking of structural proteins (44, 45); ABCA12, encoding a transporter involved in lipid transport (46); STS, which is responsible for the metabolism of cholesterol sulfate; and ALOX12B [encoding arachidonate 12-lipoxygenase (12R-type)], ALOXE3, and CYP4F22, each of which is involved in the arachidonic acid cascade (7, 47–49). Mutations in ALOX12B, ALOXE3, and CYP4F22 are associated with ichthyosis, and mice with a null mutation of the Alox12b gene have red, scaly skin and die prenatally. Putative roles of ALOX12B, ALOXE3, and CYP4F22 in the metabolism of arachidonic acid are depicted in Figure 6.
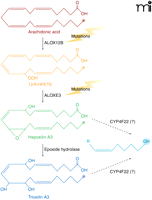
Steps where genetic mutations are linked to ichthyoses are illustrated. Mutations in either ALOX12B or ALOXE3 result in nonbullous congenital ichthyosiform erythroderma (NCIE), also referred to as type 5 lamellar ichthyosis. We speculate that CYP4F22 could modulate hepoxilin/trioxilin bioactivity in type 3 lamellar ichthyosis as indicated. (The omega position is highlighted in each case, suggesting a role for CYP4F22.)
Like CYP4V2, CYP4F22 is considered to be an “orphan” member of the CYP4 family (50) with no previously ascribed function. CYP4F22 is most closely related to CYP4F2 and CYP4F3, with 67% and 66% protein sequence identity, respectively. CYP4F22 shares 37% sequence identity with CYP4V2. The identification of mutant CYP4F22 as the causative gene for type 3 lamellar ichthyosis was made in 2006 following genetic analysis of twenty-one patients from Algeria, France, Italy, and Lebanon (7). It is interesting to note that among all of the affected individuals, representing a total of only seven distinct mutations, each was homozygous for the disease-associated allele. The seven lamellar ichthyosis–associated mutations of CYP4F22 represented five amino acid substitutions (i.e., F59L, R243H, R372W, H456Y, and H436D), one frame-shift mutation, and one large deletion resulting in loss of exons 3–12 (Figure 2B). The latter two mutations would clearly be expected to abrogate CYP4F22 catalytic activity, and it seems likely that the nonconservative nature of the homozygous coding-region mutations also could adversely affect CYP4F22 function. However, as is the case in BCD, no experimental studies have yet been conducted with any mutant forms of CYP4F22. Mapping of the five coding-region mutations sites to a homology model for CYP4F22 indicates that only H435 is buried in the enzyme’s core, whereas F59, R243, and R372 are solvent-exposed; H436 also appears to be solvent-accessible (Figure 3). Therefore, as in the case of disease-associated CYP4V2 mutations, it seems likely that multiple mechanisms could underlie any functional deficits in enzyme activity attributable to these nonsynonymous mutations.
Trioxilins and Hepoxilins as Potential CYP4F22 Substrates
Based, in part, on analogy to the leukotriene lipoxygenase pathway and knowledge that other lamellar ichthyosis–associated genes (ALOX12B and ALOXE3) had functions within the hepoxilin lipoxygenase pathway, it has been proposed that a physiological substrate of CYP4F22 is trioxilin A3 (7). However, in the five years since the initial identification of CYP4F22 variants in lamellar ichthyosis, there is only a single report investigating the catalytic function of recombinantly expressed CYP4F22 (51). This study employed a yeast expression system to demonstrate that recombinant CYP4F22 could oxygenate arachidonic acid (20:4 n-6) at C-18, indicative of fatty acid ω-3 hydroxylase activity as opposed to ω-hydroxylation. However, this CYP4F22-dependent activity was one to two orders of magnitude lower than that observed for a different CYP4 enzyme, CYP4F8, and so the significance is unclear (51). If CYP4F22 functions within the 12R-lipoxygenase pathway, the proposal has been that it might catalyze hydroxylation of trioxilin A3 to 20-hydroxy trioxilin A3 (7). 20-Hydroxy trioxilin A3 could then be further oxidized to 20-carboxy-(R)-trioxilin A3 (20-COOH TXA3) by ALDH3A2 and ichthyin (7). Unfortunately, the biological relevance of trioxilin or any related ω-hydroxylated products to skin physiology is unknown.
On the other hand, there does appear to be both genetic and biochemical evidence to support a key biological role for hepoxilins in the formation of the water barrier of the epidermis (52). Hepoxilins exert a wide variety of biological effects including stimulation of intracellular calcium levels, insulin secretion, and vascular permeability. With regard to vascular permeability, the effects of hepoxilins on rat skin have been demonstrated in vivo with evidence for epimer-specific changes (53). Although the barrier abnormality of lamellar ichthyosis could be related to deficiencies in essential fatty acids in the epidermis, a more plausible explanation may lie in alterations in hepoxilin signaling. It has been shown that epidermal lipoxygenase-mediated hepoxilin products selectively activate the nuclear receptor peroxisome proliferator-activated receptor (PPAR) α (54). Epidermal activation of PPARα causes a variety of effects associated with eicosanoid signaling, including stimulation of anti-inflammatory and pro-differentiating activities (55, 56). Similar to the situation described above for certain docosanoids, human neutrophils can ω-hydroxylate hepoxilin A3, although this activity appears to be independent of the leukotriene B4 hydroxylase CYP4F3A (57). In preliminary experiments, we have found that human leukocytes express CYP4F22 mRNA (P. Rohatgi and E.J. Kelly; unpublished observation). Thus, deficits in hepoxilins and/or their metabolites are clearly associated with lamellar ichthyosis and CYP4F22-dependent ω-hydroxylase activity may be key to normal keratinocyte function. With this in mind, understanding the hepoxilin/trioxilin substrate specificity of CYP4F22 as well as the enzyme’s tissue localization and activity in normal and diseased skin are essential components to defining the biochemical relationship between CYP4F22 mutations and lamellar ichthyosis.
Concluding Remarks
The seemingly disparate diseases of BCD and lamellar ichthyosis and their linkage to members of the CYP4 family provide an invaluable opportunity for investigating the normal physiological functions of these enzymes. Both P450s are likely involved in maintaining normal lipid homeostasis in their target organs. It is also interesting to note that whereas BCD patients are not known to manifest any symptoms that significantly overlap with symptoms of patients with lamellar ichthyosis, there are other diseases that exhibit retinitis pigmentosa/ocular crystal deposition and ichthyoses. Patients afflicted with either Sjögren–Larsson syndrome or Refsum’s disease present with comorbidities identical in appearance to BCD and lamellar ichthyosis (58, 59). Thus, one might conclude that the ALDH3A2 and PHYH gene mutations associated with Sjögren–Larsson syndrome and Refsum’s disease, respectively, are intersecting downstream of the eicosanoid-signaling pathways perturbed by mutations in CYP4V2 and CYP4F22. Although these CYP4 enzyme mutations may simply be altering lipid membrane architecture, a more attractive hypothesis evolves from the defining characteristic of CYP4 enzymes, fatty acid ω-hydroxylase activity. Much as CYP4F3A modulates the chemotactic/inflammatory properties of leukotriene B4, CYP4V2 and CYP4F22 may be regulating the function of eicosanoid (specifically docosanoid and hepoxilin) signaling molecules, respectively. Admittedly speculative in nature, these conclusions provide a framework for future studies on the biochemical mechanisms underlying these fascinating cytochrome P450–linked diseases.
Acknowledgments
This work was supported in part by the National Institutes for Health [Grant GM49054 (AER)] and by a Drug Metabolism, Pharmacogenetics and Transport Research grant (EJK) from the School of Pharmacy, University of Washington.
Footnotes
-
Authorship contributions
Wrote or contributed to the writing of the manuscript: Kelly, Nakano, Rohatgi, Yarov-Yarovoy, and Rettie.
- Copyright © 2011
References
Edward J. Kelly, PhD, is Research Assistant Professor in the Pharmaceutics Department at the University of Washington in Seattle. Trained in molecular biology, his research focuses on generation and characterization of mouse models of human gene function, particularly cytochrome P450s. Email edkelly{at}u.washington.edu.

Allan E. Rettie, PhD, is Professor and Chairman of the Department of Medicinal Chemistry at the University of Washington in Seattle. Trained in drug metabolism, his interests pertain to the enzymology and pharmacogenetics of oxidative drug metabolism. Email rettie{at}u.washington.edu.

Vladimir Yarov-Yarovoy, PhD, is an Assistant Professor in the Department of Physiology and Membrane Biology at the University of California, Davis. Trained in biophysics and computational biology, his interests are focused on computational protein structure prediction and design.

Mariko Nakano, BS, is a PhD candidate in the Department of Medicinal Chemistry at the University of Washington in Seattle. Her PhD thesis is focused on elucidating the biochemical characteristics of CYP4V2 and its role in the ocular disease, Bietti’s crystalline dystrophy.

Priyanka Rohatgi, PhD, is a Pharmacokinetics Scientist at Baxter Healthcare, Round Lake, IL. Trained in pharmacokinetics, drug metabolism, biochemistry, and bioinformatics, her research interests pertain to pharmacokinetics and clinical pharmacology.


