Autoimmunity and Lymphoproliferation: Two Genes are Worse than One
Systemic lupus erythematosus (SLE) is a serious illness that causes widespread inflammation and organ damage. Its basis is understood to be autoimmune, yet its precise cause and much of the basis of its pathology remain unknown. Research firmly indicates that there is a genetic component to SLE pathogenesis. The disease has long been known to cluster in certain families; and for identical twins, when one has the disorder, there is a high likelihood that the other twin will also have SLE (1) . Additionally, SLE is both more prevalent and more severe in certain ethnic groups, further implicating an important genetic component in its causation.
Rapid progress has occurred in SLE genetics in recent years. Important chromosomal intervals that contain candidate genes have been defined in studies of SLE families (2) . The existence of excellent models of SLE in inbred mice has also led to important insights and to a greater appreciation of gene interactions in producing a disease phenotype. Using recombinant inbred mice originally derived from the classic SLE-prone NZB x NZW F1 cross, Wakeland and collaborators have defined a series of NZW-derived genes that act synergistically to produce features of the SLE phenotype (3) . Mutations in the Sle1 locus—containing four important genes, termed Sle1a, Sle1b, Sle1c, and Sle1d —result in a loss of tolerance to certain nuclear antigens (proteins). The functions of the products of these four genes are further modified by the participation of suppressor genes. Mutation of Sle2 leads to the diffuse activation of B lymphocytes, and expression of certain alleles at the Sle3 locus leads to a lymphoproliferative phenotype resulting from diminished apoptosis. These genes interact in an additive and epistatic fashion to produce features of SLE, such as the production of nuclear-protein–specific antibodies and inflammatory renal disease.
Fas is another important gene implicated in autoimmunity. In both humans with ALPS (autoimmune lymphoproliferative syndrome) (4) and in mice (5) , loss-of-function mutations in the apoptosis-inducing receptor Fas or in its ligand (FasL) lead to dramatic accumulation of nonmalignant lymphocytes in peripheral lymphoid organs, accompanied by systemic autoimmunity. The recessive Fas mutation in mice is known as lpr , and the recessive FasL mutation as gld. Autoimmunity in individuals with lesions in the Fas apoptotic pathway is thought to result from impaired apoptotic deletion of autoreactive peripheral B and T cells, whereas coincident lymphoproliferation reflects the accumulation of these and of many additional lymphocytes, which would normally be deleted in the course of immune responses. It is especially striking that anergic (immunologically unresponsive) cells with unusual phenotypes progressively fill lymph nodes and spleen. Most of these cells, unlike normal mature T cells, express neither CD4 nor CD8. Though they express T-cell receptors and other T-cell markers, they also bear certain B-cell markers such as the CD45 isoform B220.
The degree and nature of autoimmunity caused by Fas mutations is highly dependent on ill-defined background genes. In C57BL/6 mice, for example, the lpr mutation causes mild autoimmunity, whereas MRL mice containing a mixed genetic background develop a fulminant lupus-like disease accompanied by vasculitis when they inherit the lpr Fas mutation (6) . In a recent article, Mohan and colleagues give insight into interactions between Fas mutations and NZW-derived SLE genes (7) . They report that Sle1 , when expressed even hemizygously in Fas-deficient C57BL/6/lpr mice, causes massive lymphoid enlargement and a robust SLE phenotype. These mice develop high levels of nuclear-protein–specific antibodies and a proliferative glomerulonephritis very similar to that observed in SLE. Prominent among the expanded cell populations in these animals are B1 B cells, which are found in the peritoneal cavity and produce certain autoantibodies, and CD4+ T helper cells. These results raise interesting questions not only related to the genesis of SLE, but also to the role of apoptotic pathways in the homeostasis of the normal immune system.
Regulation of the Lymphocyte Pool
Both T and B cells undergo rigorous negative selection early in their development, so that most autoreactive lymphocytes are eliminated through apoptosis before reaching the mature lymphoid compartment. Further negative selection proceeds even among mature cells, assuring that self-reactive and potentially disease-causing lymphocytes are removed, together with senescent or superfluous cells. Simultaneously, positive selection for useful lymphocytes occurs in newly maturing cells, mainly in the thymus and liver, and among fully differentiated lymphocytes during immune responses.
How do these SLE-causing genes interact to cause the formation of autoantibodies? Although it remains unknown which genes within the Sle1 locus promote lupus, one component at least (Sle1b ) is expressed in B cells (8) , and it is likely that other genes within this locus also encode molecules of fundamental importance in the regulation of lymphocyte activation. These aberrant B (probably with the help of autoreactive T) cells produce histone-specific antibodies; however, the resulting autoimmunity falls short of a full SLE syndrome. The present study (7) argues strongly that it is activation-induced cell death (AICD)—mediated through Fas—that limits both B and T cell activation, causing selective apoptosis of activated lymphocytes. Very likely this occurs after the activation of mature B and T cells in the peripheral immune system, because lymphocytes in lpr mice undergo normal, intrathymic negative-selection (Box 1). The data also emphasize that multiple apoptotic pathways are used for the elimination of autoreactive lymphocytes. Although the degree of functional overlap between Sle3 and Fas was not directly addressed, findings imply that certain lymphoid subsets may rely more heavily on one apoptotic pathway than on another.
These data emphasize the complexity inherent in the potential interaction of genes that regulate the threshold of B-cell activation (CD19, CD22, Lyn, and SHP-1, for example) (9,10) , and the variety of genes that might mediate apoptosis [Fas, CD27, and several members of the tumor necrosis factor (TNF) receptor gene family]. Stimulation of one or more clones of autoreactive T or B cells may be insufficient to trigger autoimmunity because of the safeguard of regulatory apoptotic mechanisms, such as negative selection. The failure of negative selection to delete even one autoreactive T or B cell clone may underlie a lifelong risk for autoimmunity should autoreactive stimuli (from viruses, drugs, or environmental agents) activate autoreactive lymphocytes. Such activation need not require an exogenous agent, but could instead come from stochastic events leading to dysregulation of B or T cells.
These genetic findings have implications for the regulation of lymphoproliferation as well as for the control of autoreactivity. Notably, the degree of lymphoid enlargement induced by the lpr mutation in the Fas gene is much greater when the disease-associated allele of Sle1 is also expressed. Although lpr -induced lymphoproliferation was known to be dependent on background genes, Sle1 is the first such locus to be defined. This finding supports the view that the accumulation of lymphocytes is driven by their state of activation, presumably driven by the presence of activating immunogen, and is dependent on genes that regulate the response to the antigen (Figure 1⇓). This view is consistent with the reduction in T-cell lymphoproliferation in lpr mice with defective or absent B cells, which results in global impairment in antigen presentation. It is important to note that the antigenic stimulus driving the lymphoproliferation need not arise from environmental conditions, because lpr mice raised under germ-free and antigen-free conditions can also have lymphoid enlargement nearly equivalent to that observed in animals housed in conventional facilities (11) .
Even within ALPS families, in which affected members share the same dominant negative Fas mutation, it has been puzzling that certain individuals have minimal lymphoproliferation while others have dramatic expansion of their peripheral lymphoid compartment. The epistatic interaction of Fas and Sle1 or similar genes could explain the variability of the ALPS phenotype. The data from mouse studies imply that the net result of the interactions of other gene products that affect lymphocyte activation ultimately results in the ALPS phenotype. Nonmalignant lymphoproliferation is also seen in Sjögren’s syndrome (12) and in certain cases of HIV infection (13) , although the cellular composition of these conditions differs markedly. These and other lymphoproliferative disorders may also require two or more genetic “hits.”
SLE genetics is advancing at a rapid pace. In the near future, we should expect to see further clarification of the function of Sle1 -encoded proteins and how they interfere with immune tolerance. It should also soon become clear how it is that these aberrant genes interact with apoptosis-controlling genes to result in severe autoimmune disease and lymphoproliferation. An increased understanding of these mechanisms may lead to better therapy both for lymphoproliferation and autoimmunity.
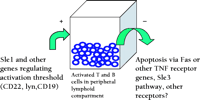
Regulating activated peripheral T and B cells bySle1 and possibly other genes. The size of this compartment is also controlled by activation-induced cell death (AICD) through Fas, wild-type Sle3, or other apoptosis-mediating receptors. Factors favoring the accumulation of cells (designated “+”) lead to the entry of additional cells into the lymphoid pool (left arrow). Those genes promoting reduction in cell numbers (designated “–”) result in a smaller pool (right arrow).
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Philip L. Cohen, MD , is Professor of Medicine at the University of Pennsylvania School of Medicine. He acknowledges the NIH, the Department of Veterans Affairs, and the Lupus Research Institute for support of his research into mechanisms of autoimmunity. Email philipco{at}mail.med.upenn.edu

