Aurora-A Overexpression Leads to Override of the Microtubule-Kinetochore Attachment Checkpoint
The aurora proteins are kinases that belong to a recently characterized family first isolated in S. cerevisiae (1). Whereas yeasts possess only one aurora, the genomes of both C. elegans and D. melanogaster encode two auroras (aurora-A and -B), and mammalian genomes code for three (aurora-A, -B and -C) (2 –5). Aurora-A, a centrosome-localized mitotic kinase involved in spindle assembly and stability, phosphorylates proteins such as Eg5 (a kinesin-related protein) (6), TACC (transforming acidic coiled-coil protein) (7), and TPX2 (targeting protein for Xklp2) (8). In addition to being a substrate, TPX2 triggers the autophosphorylation (and activation) of aurora (9, 10).
The AURORA-A gene is located at chromosome position 20q13, a site frequently amplified in breast and colorectal tumors (11). Overexpression of aurora-A (together with aurora-B and -C) has been observed in many tumor cells. In addition, overexpressed aurora-A can transform Rat1 and NIH3T3 cells (12, 13). Several phenotypic and genotypic changes have been observed after aurora-A overexpression in culture cells: abnormal mitosis, polyploidy, centrosome amplification, and abrogation of the DNA damage-induced G2 checkpoint. In a recent paper, Anand and collaborators also reported that overexpression of aurora-A can override the mitotic checkpoint (14). But which of these events is involved for the appearance of the transformed phenotype? Most likely, all are necessary, although none is independently sufficient. Overexpression of active or inactive aurora-A always provokes aneuploidy and centrosome amplification, a phenotype observed in vivo in many tumor cells (12, 13, 15, 16), suggesting that the kinase activity of aurora-A is not essential for these effects. It was thought that both events might lead to genomic instability and tumorigenesis, and consequently, establish the early steps in the tumorigenesis process (17, 18). However, this phenotype alone is not sufficient to explain the oncogene potential of aurora-A because only the active kinase transforms cells.
In order to understand how aurora-A can transform cells, the efficiency of mitotic checkpoints has been analyzed in cells overexpressing the kinase. For instance, the inhibition of aurora-A by microinjection of aurora-A-specific antibodies into cells delays mitotic entry, whereas overexpression of aurora-A results in premature entry into mitosis in the presence of DNA damage induced by X-irradiation (19). These results suggest that aurora-A overexpression abrogates the DNA damage-induced G2 checkpoint. [This function has already been attributed to the single aurora in yeast (20). Until now it was thought that the mammalian aurora-B protein that localized at the kinetochore was the most likely aurora candidate for involvement in the spindle checkpoint (21).]
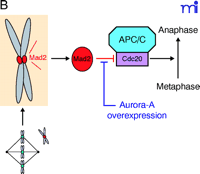
Now a new player has emerged to resolve the mystery. When the checkpoint is normally activated, cell cycle progression arrests in metaphase with Mad2 (mitotic arrest deficient 2) remaining localized at the unattached kinetochores. Anand and collaborators report that in cells overexpressing aurora-A, even with Mad2 decorating kinetochores, the metaphase arrest is not maintained (14) (Figure 1A⇓). This result suggests that in the presence of an excess of aurora-A the activated mitotic checkpoint signalled by Mad2 is abrogated. Mad2 functions as an inhibitor of Cdc20, an activator of APC/C (anaphase promoting complex or cyclosome) that participates in the degradation of proteins and thereby triggers the metaphase-anaphase transition. The presence of Mad2 at any kinetochore during mitosis signals to the cell the presence of an unattached chromosome that precludes the metaphase-anaphase transition. As soon as the kinetochore is captured by microtubules, Mad2 dissociates from kinetochores; the inhibition of Cdc20 is relieved and APC/C becomes active. The data from Anand and collaborators suggest that aurora-A, when overexpressed, acts downstream of Mad2 and upstream of Cdc20 to override the mitotic checkpoint (Figure 1B⇓). This is interesting because although aurora-A is degraded by the APC/C in a Cdh1-dependent manner, the kinase has previously been reported to associate with Cdc20 (16, 22, 23). Accordingly, the physiological signification of the Cdc20–Aurora-A association remains to be elucidated. Also, whether aurora-A phosphorylates Cdc20 remains unanswered.
Aurora-A is recognized as an oncogene because its kinase activity can transform NIH3T3 cells (13). Anand and collaborators choose to overexpress aurora-A in primary cell culture in order to assay its transformation potential. They were not able to transform the cell lines, indicating that aurora-A needs a particular background of abnormalities for its transformation activity to be revealed. Thus, research should be directed toward identifying the additional factors necessary for overexpressed aurora-A to trigger cell transformation. In fact, this was already implied by the observation that only two cell lines have been successfully transformed by aurora-A overexpression (12, 13). Anand and collaborators also report that, compared to chromosomes in immortalized cells, the chromosomes in primary cells expressing catalytically inactive aurora do not become polyploid (Figure 2⇓) (14). However, whether overexpression of inactive aurora-A also results in mitotic checkpoint override has yet to be elucidated.
Anand et al. also report that whereas HeLa cells treated with taxol arrest in metaphase and eventually die by apoptosis, HeLa cells overexpressing aurora-A become resistant to apoptosis. This compelling result should have an extremely important impact: taxol is a drug currently used in several chemotherapeutic cancer protocols and aurora-A is overexpressed in cells from various tumors. Several clinicians have already noticed that in some cases lowering taxol in chemotherapy increases the efficiency of the cancer cure.
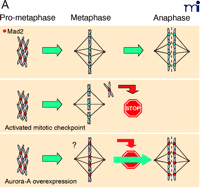
Mad2 binding to kinetochores during metaphase-anaphase transition in HeLa cells. A. (Top)
In prophase every kinetochore is decorated with Mad2. Once a kinetochore is captured by microtubules, Mad2 labelling disappears. Metaphase-to-anaphase transition will be triggered only when all kinetochores are captured. However, any kinetochore remaining decorated with Mad2 (i.e., unattached chromosome) will activate the mitotic checkpoint leading to cell cycle arrest (Middle). In the presence of overexpressed aurora-A, the cell ignores the Mad2 signal and triggers metaphase exit (Bottom). B. Mad2 is an inhibitor of Cdc20, a known APC/C activator. Overexpression of aurora-A interferes with the Mad2–Cdc20 signal that promotes progression to anaphase (14).
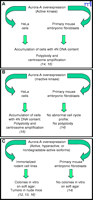
Effect of overexpression of various forms of aurora-A in different cell lines. A. Overexpression of active aurora-A in immortalized or primary cells leads to polyploidy and centrosome amplification. B. Overexpression of inactive aurora-A in primary cells does not lead to polyploidy and centrosome amplification. C. Overexpression of active, hyperactive, or nondegradable aurora-A transforms immortalized cells but not primary cells. Associated references in parentheses.
Acknowledgments
Dr. Prigent’s research is financed by the “Ligue Nationale Contre le Cancer” (équipe labellisée).
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Claude Prigent, PhD, is Director of Research at the CNRS (Centre National de la Recherche Scientifique), and head of the Cell Cycle Group, at the UMR6061 Genetics and Development, in the University of Rennes 1, France. Address correspondence to CP. Email: claude.prigent{at}univ-rennes1.fr

Stéphanie Dutertre, PhD, is currently a postdoctoral fellow with Claude Prigent at the UMR6061 Genetics and Development in the University of Rennes 1, France. She is working on the relationship between aurora-A and oncogenesis. She is a fellow of the ARC (Association pour la Recherche sur le Cancer). Previously, her doctoral studies with Mounira Amor-Gueret focused on the function and regulation of the helicase BLM, mutated in Bloom syndrome. Email stephanie.dutertre{at}univ-rennes1.fr

