- Institution: Stanford Univ Med Ctr Lane Med Lib/Periodical Dept/Rm L109
- Sign In as Member / Individual
LQT4 Gene: The “Missing” Ankyrin
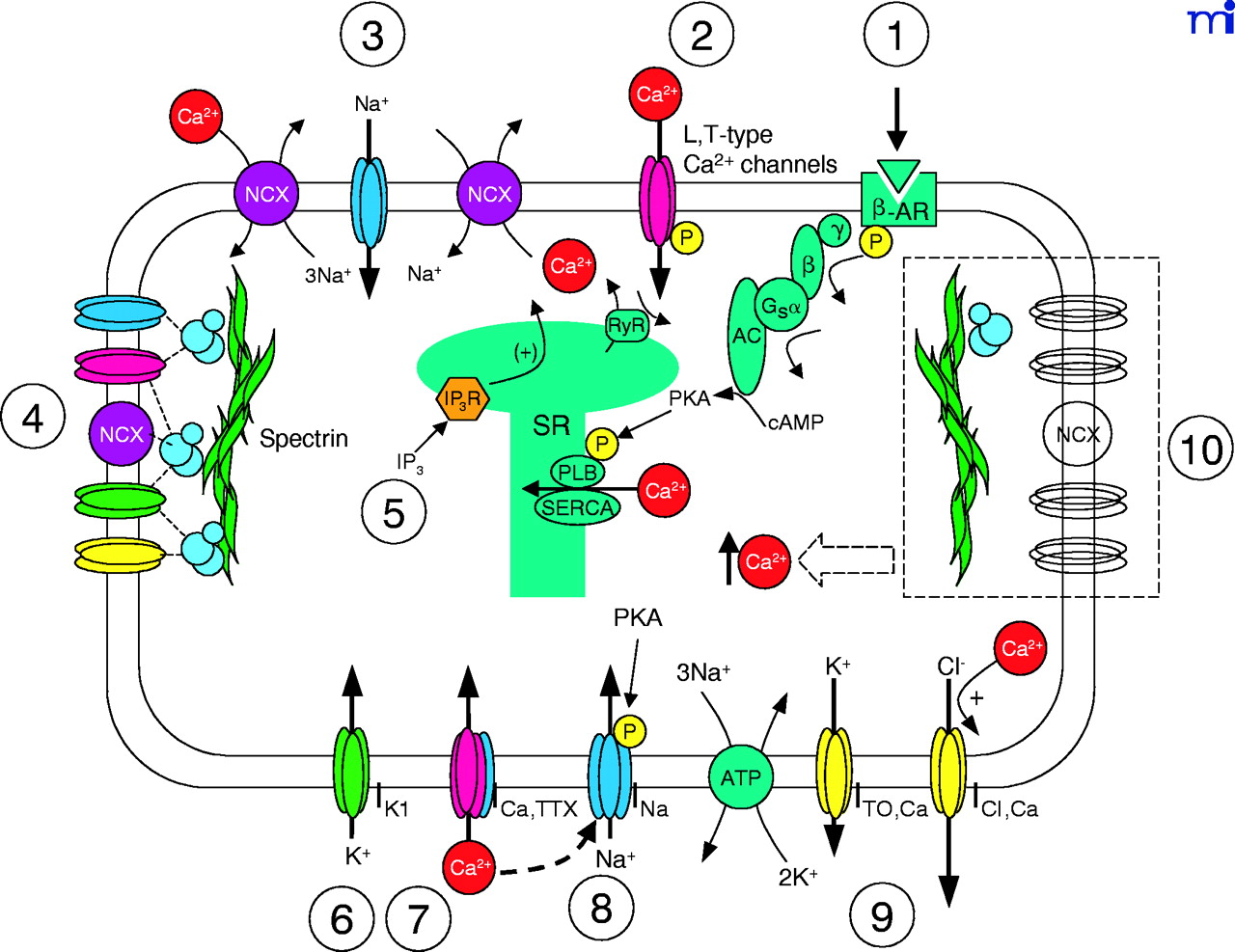
Candidate mechanisms for intracellular Ca2+handling in ventricular myocytes. The proposed mechanisms are as follows: (1) β-adrenergic receptor (β-AR) stimulation, via Gs-mediated activation of adenylate cyclase (AC) and protein kinase A (PKA), phosphorylates phospholamban (PLB) to relieve PLB’s inhibition of the Ca2+-ATPase pump (SERCA) and, thereby, to enhance Ca2+ uptake by the sarcoplasmic reticulum (SR). (2) Ca2+-induced Ca2+-release (CICR) through the ryanodine receptor (RyR) is mediated by L-(or T-) type Ca2+ channel activation. (3) CICR via activation of voltage-dependent INa and the dual roles of the Na+/Ca2+ exchanger (NCX). (4) Ankyrin-mediated (blue spheres) localization of ion transport proteins via spectrin binding. (5) Ca2+ release triggered by inositol -1,4,5-trisphosphate (IP3) through IP3 receptors (IP3R). (6) Inward rectifying K+ current (IK1) maintains resting membrane potential and is sensitive to changes in [Ca2+]i homeostasis. (7) CICR triggered by Ca2+ entry through tetrodotoxin (TTX)-sensitive Ca2+ channel. (8) Ca2+ release mediated by “slip mode conductance”, in which PKA-dependent phosphorylation of TTX-sensitive INa have altered permeability for Ca2+. (9) Ca2+-insensitve (ITO,Ca) and Ca2+-sensitive (ICl,Ca) transient outward potassium channels, the latter of which is carried by Cl− rather than K+ ions. (10) Reduced Ca2+-handling proteins due to a deficiency of ankyrin (blue spheres).

