Phosphorylation of G Protein–Coupled Receptors: GPCR Kinases in Heart Disease
- 1Department of Surgery,
- 2Department of Pharmacology,
- 3Department of Cancer Biology, Duke University Medical Center, Durham, NC 27710
- Address correspondence to WJK. E-mail koch0002{at}mc.duke.edu; fax 919-684-5714.
Abstract
In the heart, β -adrenergic receptors (β ARs), members of the superfamily of G protein–coupled receptors (GPCRs), modulate cardiac responses to catecholamines. β AR signaling, which is compromised in many cardiac diseases (e.g., congestive heart failure), is regulated by GPCR kinases (GRKs). Levels of the most abundant cardiac GRK, known as GRK2 or β AR kinase 1 (β ARK1), are increased in both animal and human heart failure. Transgenic mouse models have demonstrated that β ARK1 plays a vital role in cardiac function and development, as well as in the regulation of myocardial signaling, and pharmacological studies have further implicated GRKs in the impairment of cardiac GPCR signaling. Gene therapy, along with the development of small-molecule modulators of GRK activity, has indicated in multiple animal models that the manipulation of GRK activity may elicit therapeutic benefits in many forms of cardiac disease.
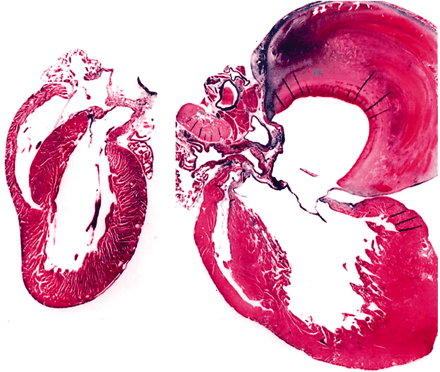
Compared to a normal mouse heart (left), the overexpression of human β2-adrenergic receptors in transgenic mice results in morbid hypertrophy (right). The regulation of β-AR activity in the heart
represents an important therapeutic target.
Image adapted with permission from SB Liggett et al. Early and delayed consequences of β2-adrenergic receptor overexpression in mouse hearts. Circulation 101, 1707-1714 (2000).
Introduction
Heart disease is the leading cause of death and illness in the United States, and hospitalizations secondary to heart failure (HF) are dramatically increasing (1). As the pathological endpoint in many cases, congestive HF is characterized by a loss of cardiac function and inotropic reserve. The sympathetic nervous system, which is a critical regulator of cardiac function, has been implicated in the inability of the failing heart to respond to stress or injury. In response to stress, catecholamines (i.e., the sympathetic neurotransmitter norepinephrine and the adrenal hormone epinephrine) bind to myocardial adrenergic receptors (ARs). These receptors, which include β1 - and β2 ARs, modulate cardiac function by coupling to and activating G proteins, and thus belong to the large superfamily of G protein–coupled receptors (GPCRs). Under normal conditions, catecholamines bind to β ARs in the heart and activate a cascade of intracellular signaling pathways, leading ultimately to increased chronotropy and inotropy. The catecholamine agonists initiate this series of events by binding to the membrane β AR and causing it to undergo a conformational change that results in its coupling with the Gs protein. The heterotrimeric Gs protein consequently dissociates into two subunits, the GTP-binding Gα subunit that stimulates adenylyl cyclase (hence the designation Gs ), and the Gβγ heterodimer, also involved in downstream effector activities that regulate cardiac function (2) .
Under certain conditions, however, agonist binding to β ARs and other GPCRs leads to receptor desensitization and downregulation, thereby decreasing receptor signaling (3) . This process of desensitization is initiated by a family of serine/threonine kinases known as GPCR kinases (GRKs). GRKs phosphorylate the intracellular portions of agonist-occupied GPCRs, which allows a second family of molecules, the β -arrestins, to prevent further G protein coupling. Desensitization may be an adaptive response to GPCR stimulation, but can also lead to pathological loss of receptor signaling.
Phosphorylation of β ARs by GRKs in the heart is thus a critical determinant of cardiac function and has been implicated in many pathological states. Over the past decade, much attention has been focused on β AR kinase 1 (β ARK1, or GRK2), which is the most abundant GRK in the myocardium (4) . In animal models of HF (5–8) as well as in the human disease (9–11) , β ARK1 expression and activity are increased and have been implicated in β AR desensitization and compromised cardiac function. Using transgenic mouse models, the roles of β ARK1 and other GRKs in cardiac development, function, and disease have been further delineated (12) . Subsequently, several pharmacological and gene therapy strategies have been used as novel potential therapies to target GRKs in HF.
General Characteristics of Cardiac GRKs
The GRKs belong to a family of protein kinases that comprises seven distinct members. GRKs contain a central catalytic domain that is homologous to other serine/threonine kinases, flanked by N- and C-terminal domains that contain specific regulatory sites (Figure 1⇓) (13–16) . GRKs are primarily cytosolic and demonstrate a high degree of substrate selectivity in phosphorylating agonist-occupied GPCRs. GRKs can be classified by localization, substrate specificity, or mechanism of action. Two GRKs, GRK1 (or rhodopsin kinase) and GRK7, are primarily located in the retina, whereas the other five are expressed throughout the body, including the heart (13, 15) . In addition to β ARK1 (or GRK2), GRK3 (or β ARK2) and GRK5 have been studied extensively in the heart (17) . Of these three GRKs, each has been shown to phosphorylate the most common myocardial β AR subtype (i.e., β1 -AR) (18, 19) .

Domain structure of the GPCR kinases (GRKs). The central catalytic domain that typifies GRKs is similar to catalytic sequences from other serine/threonine kinases. The N- and C-terminal domains appear to determine the specific interactions between any given GRK and other proteins and components of the cell membrane. Of particular note is the C-terminal domain, which governs interaction with the Gβγheterodimer released upon activation G proteins. The C-terminal domain of βARK1 (i.e., of GRK2), denoted here as βARKct, has been transgenically expressed with therapeutic efficacy in a variety of cardiomyopathic models. See text for details.
Despite their functional similarities, β ARK1, GRK3, and GRK5 are distinct enzymes with unique regulatory mechanisms. β ARK1 and GRK3 are 85% similar in consideration of their overall primary structure, and 95% similar with respect to their catalytic domain (20) . Their similarity with respect to their C termini, however, is only 52% (21) , which accounts for their differential affinities for specific GPCRs. Localization of both β ARK1 and GRK3 at the plasma membrane is accomplished by interaction between the 200-residue C terminus of the GRK and the βγ heterodimer (Gβγ ) that is released from the G protein upon activation. The same C-terminal domain associates, in a regulated manner, with phosphatidylinositol 4,5-bisphosphate (PIP2 ) contained in the plasma membrane (22–24 ). Whereas GRK5 is constitutively bound to the membrane, the association of β ARK1 and GRK3 with the plasma membrane, as evidenced both in vitro (21, 25, 26) and in vivo (27–29) , is determined by the GPCR present. The constitutive interaction of GRK5 with the plasma membrane, on the other hand, depends on a PIP2 -binding sequence in the N-terminal domain (30) . Studies in transgenic models have shown that GRK5 in the heart consequently has a predilection for GPCRs that is distinct from that of β ARK1 or GRK3 and therefore allows for the desensitization of distinct receptors (29, 31) . The mechanisms for binding with the plasma membrane and/or with individual G proteins thus provides unique bases for GPCR selectivity among β ARK1, GRK3, and GRK5.
GRKs in Cardiac Disease
In human HF, impaired β AR signaling compromises cardiac sensitivity to inotropic stimulation (32) . The loss of receptor signaling is associated with an approximate threefold elevation in myocardial β ARK1 expression and GRK activity (9, 10) . Myocardial ischemia and hypertension have also been associated with increased expression and activity of β ARK1 (6, 11). These aspects of human heart disease are similarly evident in animal models, where β ARK1 levels are increased in cardiac hypertrophy (5) , ischemia (6) , and HF (7, 8, 33–36) . Given the variety of pathological insults represented in the animal models, β ARK1 upregulation appears to be an early common event in the pathogenesis of HF. In fact, β ARK1 elevation often precedes the development of clinical HF (34, 35) and may represent a novel early marker for cardiac dysfunction. Like β ARK1, GRK5 expression and activity are elevated in animal models (33, 37, 38) , although its role in human HF remains unclear. In contrast, GRK3 expression is not increased in human HF (10) .
GRK Alterations in Transgenic Mice
To further determine the role of GRKs in the heart, their expression and activity have been investigated in transgenic mouse models. Using the promoter from the gene that encodes the murine α -myosin heavy chain (α MHC) (39) , it is possible to overexpress specific GRK-encoding transgenes in the myocardium. Mice have been engineered with myocardial overexpression of β ARK1 (27) , GRK3 (28) , and GRK5 (31) . Alternatively, transgenic techniques have been used to create knockout mice in which the genes encoding GRK are ablated (15, 40, 41) . The resulting biochemical, physiological, and histological analyses of these transgenic mice provide powerful insights into the roles of individual GRKs in cardiac development and signaling.
Under control of the promoter from the α MHC-encoding gene, β ARK1 was overexpressed in the hearts of mice to the levels seen in human HF (27) . In these mice, left ventricular contractility in response to the β AR-agonist isoproterenol (ISO) was significantly decreased. In addition, the response to the GPCR agonist angiotensin II (Ang II) was attenuated (31) . These studies demonstrated for the first time that myocardial overexpression of β ARK1 results in β AR desensitization in vivo. In contrast, a second mouse line, overexpressing only the C-terminal domain of β ARK1 (i.e., β ARKct), exhibit enhanced left ventricular contractility and relaxation under both basal and ISO-stimulated conditions (27) . In this second line, the overexpressed C-terminal domain, containing the Gβγ -binding domain (Figure 1⇑), ultimately functions as an inhibitor of β ARK1 activity by competitively preventing Gβγ from positioning native β ARK1 at the cell membrane (Figure 2⇓) (23) . These findings demonstrate that changes in β ARK1 levels significantly influence cardiac performance.

The nexus of catecholamine receptor, G protein, and GPCR kinase: An opportunity for cardiotherapeutic intervention. Schematized is a molecule of βARK1 (i.e., GRK2), the most abundant cardiac GRK, which is positioned, through the interaction between its C-terminal domain and the βγ heterodimer (Gβγ) of an activated G protein, so as to phosphorylate a βAR molecule. Phosphorylation results in cellular desensitization to catecholamines, which may be involved is certain pathological conditions. To prevent the phosphorylation of the βAR (and thereby preclude desensitization), therapeutic strategies may utilize peptides or small molecules that interfere with the interaction between GRKs and the Gβγ heterodimer.
To investigate the role of GRKs in cardiac development, additional knockout mouse lines have been studied, each of which lacked a GRK. Embryos devoid of the β ARK1-encoding gene develop major cardiac anomalies and fail to survive past fifteen days of gestation (40) . In contrast, knockout mice lacking either GRK3 or GRK5 were viable without any observable alteration in cardiac function (15) . Subsequently, heterozygous β ARK1 knockout mice (Adrbk1+/-) , expressing 50% less β ARK1 than wild-type animals, were found to have enhanced β AR signaling and contractility similar to the β ARKct-expressing mice (41) . Going one step further, the β ARKct-expressing and Adrbk1+/- lines were crossed, resulting in mice demonstrating the least amount of GRK activity and the most enhanced cardiac contractility (41) . These studies may suggest that of the three GRKs studied in the heart (i.e., β ARK1, GRK3, and GRK5), β ARK1 is the most important for myocardial development and function.
To address the in vitro selectivity of GRKs for specific GPCRs, lines of mice were created with cardiac overexpression of either GRK5 or GRK3 (31, 28) . As in the case of β ARK1 overexpression, mice with cardiac overexpression of GRK5 exhibit a marked loss of β AR signaling and inotropic reserve (31) ; however, cardiac response to Ang II was not concomitantly altered, providing in vitro evidence for differences in substrate selectivity among different GRKs. GRK3-overexpressing mice, moreover, have normal cardiac function and β AR signaling (28) , but exhibit decreased signaling through the thrombin receptor pathway, confirming previous in vitro studies (26, 42) . These findings further call into question the traditional assumption that β ARK1 and GRK3 are isozymes.
Regulation of βARK1 in Normal and Failing Myocardium
Clearly, both knockout and transgenic mouse models confirm the importance of β ARK1 expression and activity in the regulation of cardiac physiology. Its upregulation in human HF, furthermore, suggests an association between β ARK1 and the pathogenesis of cardiac dysfunction. Because neurohormonal activation also occurs early in the progression to HF, as is reflected by increased catecholamine levels and adrenergic drive immediately after myocardial infarction (MI) and long before progression to end-stage HF (43, 44) , sympathetic nervous activity has been investigated as a possible trigger for increasing GRK activity in failing myocardium. Specifically, normal mice exposed to a sustained infusion of ISO manifest increased expression and activity of β ARK1 and develop myocardial hypertrophy with impaired β AR signaling (45) ; as seen in other studies, GRK5 expression is unaffected (45) . In addition, infusion of the β -blockers atenolol and carvedilol lead to decreased β ARK1 expression and enhanced β AR signaling (45) . A porcine model similarly demonstrated a reduction in GRK activity and increased β AR signaling following chronic β AR blockade (46) .
The relationship between β ARK1 and β AR-mediated cardiac dysfunction has been further defined in normal rats subjected to chronic adrenergic activation by means of a salt-deprived diet (47) . Despite the absence of discrete changes in cardiac morphology in this model, the elevated catecholamine state nevertheless led to β ARK1-mediated desensitization and β AR downregulation. Subsequently, treatment with the β -blocker atenolol reversed these biochemical changes, normalizing both β ARK1 levels and β AR density (47) . Conversely, the presence of cardiac hypertrophy alone is not associated with abnormalities in β AR signaling. Specifically, mice treated with the β1 -AR agonist phenylephrine (PE) develop cardiac hypertrophy in the absence of elevated β ARK1 levels (48) . Accordingly, the in vitro treatment of isolated myocytes with ISO but not PE induces the expression of β ARK1 (48) . The catecholamine-stimulated increase in β ARK1 expression thus appears to be a β AR-selective process that can occur in the absence of overt cardiomyopathies.
Manipulation of βARK1 in Murine Models of Heart Failure
Transgenic mice clearly identifiy GRKs as potential targets in the treatment of HF. The importance of β ARK1 as a modulator of β AR signaling is particularly underscored by the enhanced cardiac function resulting in transgenic mice that express β ARKct, a peptide inhibitor of β ARK1 activity (Figure 1⇑) (27) . The β ARKct transgene effectively rescues several models of murine HF (7, 49–52) . This strategy was first used in a mouse model of cardiomyopathy resulting from knockout of the MARCKS-like protein (Mlp-/- ) (7) . β ARKct expression in the Mlp-/- mice restored left ventricular contractility and inotropic responsiveness, thereby halting progression to HF. Similar results were obtained when mice overexpressing the Ca2+ binding protein calsequestrin (CSQ) were crossed with β ARKct-expressing transgenic mice (51) . The resulting CSQ- and β ARKct-expressing progeny had a significantly lengthened life span (and improved cardiac function) relative to the CSQ-expressing parental line (15 vs 9 weeks). Interestingly, the beneficial effects of β ARKct expression and treatment with a β -blocker in mitigating the CSQ phenotype prove to be synergistic (51) . In another study, the previously described β AR signaling defects resulting from overexpression of β ARK1 (27) , including blunted responsiveness to β -agonists as well as elevated β ARK1 activity, were ameliorated by concomitant β ARKct expression (49) . Similarly, expression of β ARKct in a cardiomyopathic mouse model carrying a mutated myosin heavy chain reverses cardiac dysfunction and hypertrophy (50) . The protective effect of β ARKct in this study was not achieved simply by increased adrenergic signaling, as overexpression of the β2 -AR transgene was unable to rescue the phenotype. Instead, these findings underlie the importance of selective reversal of β ARK1 desensitization as a potential therapeutic strategy.
In addition to rescuing genetically engineered phenotypes of murine HF, β ARKct has also been used to ameliorate several models of acquired cardiac injury. Whereas ischemia/reperfusion in β ARK1-overexpressing mice results in globally decreased cardiac function relative to a control genotype (53) , β ARKct overexpression enhanced basal cardiac function—relative to β2 AR-overexpressing mice—and better maintained cardiac function following ischemia/reperfusion (54) . In another study, transgenic mice were created with myocardial-targeted β ARKct transgene expression under control of the promoter of the gene that encodes the cardiac ankyrin repeat protein (CARP), which is activated in response to cardiac hypertrophy (55) . When adult transgenic mice were subjected to transverse aortic constriction to induce pressure overload and left ventricular hypertrophy, the resulting acute expression of β ARKct maintained β AR responsiveness and inotropic reserve. These experiments further support the use of β ARK1 inhibition as a potential therapeutic modality and laid the groundwork for in vivo studies.
Cardiac Therapeutic Potential of βARKct
Transgenic mouse models clearly identify β ARK1 as a potential target in the treatment of HF. Adenovirus-mediated gene transfer techniques represent one avenue for delivery of the β ARKct transgene to failing myocytes in vitro and to the myocardium in vivo. Myocytes isolated from failing rabbit hearts demonstrate biochemical alterations similar to those seen in human HF, including increased β ARK1 expression and activity, as well as β AR downregulation and functional uncoupling (56) . Expression of an adenovirus-delivered β ARKct transgene, however, restores β AR signaling and basal β ARK1 activity to these myocytes (56) . Similarly, failing hearts from spontaneously hypertensive rats are typified by limited contractility and relaxation and also exhibit impairment of β AR signaling and increased β ARK1 levels comparable to human HF (57) . Following adenovirus-mediated β ARKct expression, the spontaneously hypertensive rat myocytes demonstrate a significant increase in basal and ISO-stimulated β AR signaling (57) . β ARKct expression also restores ISO-stimulated cell shortening, contraction, and relaxation. These results indicate that targeted β ARK1 inhibition is possible by gene transfer and represents powerful therapeutic potential for the rescue of failing cardiomycytes.
A wide variety of in vivo animal models of HF have been developed in which to evaluate the therapeutic potential of β ARK1 inhibition. One reliable and well-characterized model is the development of HF in rabbits following MI (8) . In remarkable similarity to human HF, these failing rabbit hearts demonstrate a global reduction in β AR density accompanied by functional uncoupling. Additionally, β ARK1 expression and GRK activity are significantly elevated. These changes are associated with impaired physiological function, with decreased left ventricular contractility and relaxation, as well as elevated end-diastolic pressure (8) . This constellation of abnormalities makes this model an ideal setting in which to investigate novel therapeutic targets. When intracoronary adenovirus-mediated gene transfer was used to deliver the β ARKct transgene at the time of MI in this rabbit model, β ARK1 expression and activity were diminished, leading to preservation of β AR density and signaling (58) . These molecular changes led to preservation of in vivo cardiac contractility at baseline and in response to ISO stimulation (58) . In a subsequent study, catheter-mediated intracoronary delivery of the β ARKct transgene to the left ventricle three weeks after MI produced similar results, with restoration of systolic function and enhanced β AR-stimulated adenylyl cyclase activity (59) .
In addition to MI-precipitated HF, other models of acquired cardiac dysfunction in rabbits have been used to investigate the effects of β ARK inhibition. Selective right ventricular expression of the β ARKct transgene following pulmonary artery banding was found to improve morbidity (60) . Addressing the role of β ARK1 inhibition in the setting of cardiopulmonary bypass, global myocardial expression of β ARKct stabilizes β AR signaling and can prevent left ventricular dysfunction (61) . Expression of β ARKct in a heterotopic cardiac transplant model, moreover, ameliorated ventricular systolic and diastolic allograft function (62) . These studies confirm that in vivo delivery and expression of the β ARKct transgene is feasible and beneficial to cardiac function in a variety of pathological scenarios. See Table 1⇓ for a summary of therapeutic effects observed upon administration of β ARKct in various animal models.
Animal Models of Cardiac Dysfunction and Therapeutic Modulation of GRK Activity
Conclusions
Transgenic mouse and rabbit studies strongly suggest that the inhibition of β ARK1 activity may represent a novel approach to the treatment of human HF. In the future, therapies may exploit gene transfer techniques and the development of new drugs. A persisting area of controversy, however, revolves around the protective versus maladaptive roles that GRKs play in the heart. Traditionally, it was believed that elevated myocardial β ARK1 levels and the resulting desensitization of cardiac β ARs would be cardioprotective, and that the disruption of this “compensatory” desensitization would allow uncontrolled sympathetic nervous system stimulation by catecholamines to worsen cardiac function. Experimental models of HF, however, indicate that the inhibition of β ARK1 activity by blockade of the Gβγ interaction reverses β AR desensitization and improves cardiac performance. These results, detailed above, indeed implicate elevated β ARK1 activity and desensitization as initially protective mechanisms, but also suggest that these same mechanisms eventually become maladaptive in the failing heart (63) . This paradox has been recognized in the clinical arena, where recent evidence has demonstrated that β -blockade, and not β -agonist therapy, is beneficial in the treatment of chronic HF (64) . The apparent contradiction may be resolved by a close look at the very different mechanisms at play in augmenting β -adrenergic stimulation in the heart. Chronic β -agonist (i.e., catecholamine) stimulation of the heart clearly leads to deleterious effects, which appear to be mediated largely through β1 ARs and may be ultimately associated with apoptotic and arrhythmogenic events (63) . In contrast, direct β ARK1 inhibition as well as pharmacological β -blockade share the ability to normalize signaling through the β AR pathway by reducing receptor desensitization and GRK activity (45), preserving receptor density and enhancing catecholamine sensitivity. In this way, β -blocker therapy and β ARK1 inhibition may be complementary therapies, both acting to reduce the activity of myocardial β ARK1 and thus prevent pathological β AR desensitization (51) .
As has been discussed in this review, inhibition of β ARK1 expression and activity can be achieved in several ways. Current pharmacological therapies in HF, including β AR-antagonists and angiotensin-converting enzyme inhibitors, decrease β ARK1 expression and prevent β AR desensitization. In animal models, in vivo adenovirus-mediated expression of a peptide inhibitor of β ARK activity can be effective. Currently, efforts are underway within the pharmaceutical industry to identify small-molecule inhibitors of β ARK1 that might prove more selective and efficient at improving cardiac performance in the setting of human HF.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Walter J. Koch, PhD (right), is Professor of Experimental Surgery at Duke University Medical Center. Beginning in September of 2003, he will be the W.W. Smith Professor of Cardiology, Director of the Center for Translational Medicine, and Vice Chair for Research in the Department of Medicine at Jefferson Medical College in Philadelphia. Jonathan Hata, MD (left), is a Surgical Research Fellow at Duke Medical Center.

