Dialogue Between ERKs and JNKs: Friendly or Antagonistic?
The mitogen-activated protein kinase (MAPK) signal transduction network comprises a highly interactive series of protein kinases. These protein kinases convey, amplify, and integrate signals from the variety of diverse extracellular stimuli received at the cell’s surface. The net result may include cellular responses such as proliferation, differentiation, development, inflammation, or apoptosis. The most thoroughly characterized subgroups of the MAPK family include the extracellular signal–regulated protein kinases (ERKs), the c-Jun N-terminal kinases/stress-activated protein kinases (JNKs/SAPKs), and the p38 family of kinases. Activated MAPKs translocate to the nucleus, where they phosphorylate a variety of target transcription factors. All three groups of MAPKs are activated by dual phosphorylation at Thr-X-Tyr motifs within the activation loop of the kinase domain by one or more MAPK kinases (MAPKKs, or MKKs) also known as MAPK/ERK kinases (MEKs). MEKs are, in turn, activated by MEK kinases (MEKKs, or MAPKKKs). Thus, each MAPK cascade actually consists of three sequential kinase reactions, which are known to result in the activation of other molecules including additional protein kinases and various transcription factors (Figure 1⇓). ERKs participate in transmitting signals initiated by tumor promoters, such as phorbol esters, and growth factors, including epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) (1, 2 ). On the other hand, various forms of inflammatory signals or stress, including UV irradiation, reportedly activate JNKs and p38s (3).
The MAPK networks are commonly depicted in the literature as linear, consecutive phosphorylation cascades; however, the activation of these pathways is not mutually exclusive. For example, stresses such as heat shock and UV irradiation weakly activate ERKs and EGF slightly activates the JNK and p38 pathways (2, 4 ). In fact, in certain cells, UV strongly activates the ERKs pathway (5), and EGF induces activation of p38 (6) . In addition, many MAPKs share the same substrate specificities (7) , although the extent or amount of direct interaction among the distinct ERKs, JNKs, and p38s is not clear.
Shen et al. recently observed in COS-7 cells that prolonged activation of JNKs by overexpressed mixed lineage kinase-3 (MLK3) results in decreased EGF-mediated activation of ERK1 and 2 (8). The MLKs are a family of at least seven serine–threonine protein kinases that act as MAPKKKs (i.e., MEKKs) to activate the JNKs and p38 pathways (9). MLK3, the most well-studied family member, contains a Gly-Pro–rich N terminus and a leucine zipper region that is required for its dimerization, which, in turn, is required for MLK3-mediated activation of JNKs (9, 10 ). Additionally, MLK3 contains a Src-homology (SH3) domain, which binds to proteins containing proline-rich sequences (9, 11 ). Shen et al. (8) present data showing that expression of active MLK3 in COS-7 cells inhibits EGF- or 12-O -tetradecanoyl-phorbol-13-acetate (TPA)-induced activation of ERK1 and 2 without affecting EGF-induced Raf activation. In separate experiments, they also showed that MLK3 phosphorylated and activated MEK-1 but that MLK3 still inhibited ERK1–2 phosphorylation, leading the authors to conclude that cells expressing constitutively active MLK3 are characterized by an uncoupling of ERK and MEK activities. Further experiments indicated that inhibition of JNKs (by JNK inhibitor I or II) or MLK3 (by CEP-11004) alleviated the inhibition of ERKs by overexpressed MLK3. These results were further supported by the reversal of MLK3-induced ERKs inhibition in cells expressing active MLK3 and one of two forms of dominant negative c-Jun: TAM-67 or DBM-3. The investigators imply that expression of constitutively active MLK3 results in continual JNK activation and consequently increases Jun-mediated transcription. The chronic activation of JNKs results in the disruption of MEK1-to-ERKs signaling, leading to decreased growth factor- and mitogen-stimulated activation of ERKs.
Although interesting, these data are probably insufficient to generalize a completely friendly or antagonistic relationship between JNKs and ERKs in all cell or tissue types under all conditions; the overexpression of MLK3 is as yet of uncertain physiological significance. The JNKII inhibitor might not specifically target JNKs (i.e., JNKII inhibitor might affect other kinases’ activity as well), and the dominant negative forms of c-Jun or inhibition of c-Jun could have nonspecific effects with other transcription factors, such as retinoic acid receptors (12) or glucocorticoid receptors (13) , and may not specifically inhibit JNK–c-Jun activity.
The results of Shen et al. are partially supported by those of Black et al. (14), who found that signaling through the ERKs is markedly decreased in v-Jun–transformed chicken embryonic fibroblasts (CEFs). The deletion in c-Jun that produces v-Jun, however, totally disrupts the regulation of v-Jun by the JNKs (15). Black et al. conclude that the deactivation of the ERKs in CEFs probably was due to a multitude of effects including inactivation of ERKs by MAPK phosphatases. Shen et al. concluded that phosphatases were probably not responsible for the inactivation of ERKs in their studies, suggesting that other mechanisms might be responsible. Zhang et al. (16) reported a direct physical interaction between p38α and ERK1–2, and that the interaction results in p38α activity–dependent decreases in the phosphotransferase activity of ERK1–2. Little additional information is available supporting the general paradigm for JNK–ERK antagonism; several studies utilizing knockout cells and mice suggest that these findings are most likely cell- or tissue-type specific.
JNK knockout (Jnk–/– ) mouse cells provide a more definite answer as to the relationship between JNKs and ERKs. Wildtype or Jnk–/– murine embryo fibroblasts (MEFs) exhibited no difference in TNF-induced activation of p38 or ERKs in spite of a complete inhibition of tumor necrosis factor–α (TNFα )-induced JNKs activity in Jnk–/– MEFs (17) . In addition, TNFα -stimulated expressions of c-Jun, JunD, c-Fos, Fra1, and Fra2 proteins were markedly depressed in Jnk–/– cells. Others have observed that activation of ERKs and p38s were unaffected in Jnk1–/– bone marrow monocytes (18) . Using dominant negative (DN) mutants of JNKs, we showed that arsenic-, UVA-, UVC-, or TNF-α –induced phosphorylation of JNKs was blocked, but that phosphorylation of the ERKs (induced under the same conditions) was not affected (19–,21). In addition, DN-ERK2 blocked both UVA- or arsenic-stimulated activation of ERKs but not of JNKs (19,21). Basal levels of ERK and p38 proteins are similar in Jnk1–/– or Jnk2–/– MEFs (22) , and when Jnk1–/– or Jnk2–/– cells are exposed to UVA, phosphorylation of ERKs, but not JNKs, is induced (19). On the other hand, disruption of Jnk1 in mice has little or no effect on TPA-induced phosphorylation of ERKs (23), whereas disruption of Jnk2 appeared to attenuate the TPA-induced ERK phosphorylation in mouse epidermis in vivo (23,24).
Little doubt exists that MAPKs engage in crosstalk, but the extent or direct effect of these “conversations” is unclear. Clearly, JNKs and ERKs interact under certain conditions. The interactions have been reported to be both “friendly” and “antagonistic” and the outcome is most likely related to type of stimulus received and is cell- or tissue-type dependent.
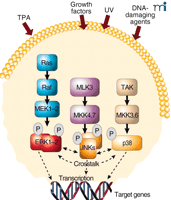
Mitogen-activated protein kinase (MAPK) signaling pathways. MAPKs are activated by a variety of stimuli, including 12-O-tetradecanoyl-phorbol-13-acetate (TPA), growth factors, UV and DNA damage. The final signal commonly includes transcription of target genes leading to cellular responses such as proliferation or apoptosis. TAK, Tumor growth factor β–activating kinase.
Acknowledgments
The authors’ work was supported by The Hormel Foundation and the National Institutes of Health grants CA 81064, CA77646, CA88961, CA27502 and CA 74916.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Ann M. Bode, PhD , is Assistant Director of the Hormel Institute, University of Minnesota; Research Associate Professor of Cellular and Molecular Biology and is a member of the Dong laboratory.

Zigang Dong, MD, DrPH , is the Hormel/Knowlton Professor and Executive Director of the Hormel Institute, University of Minnesota; Professor of Cellular and Molecular Biology. Address comments to ZD. Email: zgdong{at}hi.umn.edu; fax 507-437-9606.

