Developing a New Generation of TNFα Antagonists for the Treatment of Rheumatoid Arthritis
Abstract
The convergence of three research pathways has led to the development of a new class of biological agents that can successfully treat chronic disease, including aggressive rheumatoid arthritis (RA). Basic studies on the pathogenesis of RA identified tumor necrosis factor–α (TNFα ) as the central mediator of the chronic inflammation that results in joint swelling, degradation, and loss of function. Parallel development of monoclonal antibody (MAb) and recombinant DNA technologies has enabled the design and production of several potent biologics that can specifically block the deleterious effects of TNFα . Two MAbs (infliximab and adalimumab), as well as a receptor fusion protein (etanercept), have thus far proven safe and efficacious in pivotal RA clinical trials. These evolving technologies provide the foundation from which future biotherapies can be derived as targets are identified and validated in diverse disease states.
Role of TNFα in Rheumatoid Arthritis
Rheumatoid arthritis (RA) affects approximately 1% of the population, with an average age of disease onset of forty years. RA is a chronic, systemic, inflammatory disease characterized by joint inflammation that leads to gradual degradation of the joint. The destruction of cartilage and bone, as well as the systemic features of the disease, eventually lead to disability, unemployment, chronic illness, and premature mortality (1). Research over the past two decades has focused on identifying the biochemical mediators of this debilitating disease. While investigators were successful at cataloging many of the secreted proteins, or cytokines, that were overexpressed in the synovial fluid of RA patients, they were perplexed that multiple types of both pro- and anti-inflammatory cytokines were elevated. The puzzle was solved when synovial cells, isolated from biopsy tissue from RA patients, were maintained in culture. The addition of tumor necrosis factor–alpha- (TNFα )-specific antibodies to these synoviocyte cultures greatly reduced the production of other proinflammatory cytokines, such as interleukin (IL)-1, IL-6, IL-8, and granulocyte-macrophage colony stimulating factor (GM-CSF), an effect not seen with antibody to IL-1 (2). Further investigations confirmed that TNFα is a key mediator of RA (3). TNFα appears early in the inflammatory response, activating signal cascades within target cells and leading to secondary synthesis of cytokines, cell adhesion molecules, and inflammatory-response enzymes. In addition, TNFα perpetuates the inflammatory process by recruiting and activating leukocytes, stimulating cell proliferation, increasing prostaglandin synthesis, and stimulating bone and cartilage resorption (2). Transgenic mice that show constitutive expression of human TNFα develop erosive polyarthritis (4) . Furthermore, treatment by murine TNFα -specific antibodies in and of itself can effectively prevent the development of collagen-induced arthritis in DBA-1 mice (5,,6). Taken together, results of in vitro and in vivo research strongly suggested that anti-TNFα therapy would benefit RA patients.
As the role of TNFα in RA became apparent, the search for effective TNFα antagonists began in earnest. Attempts to identify small-molecule TNFα antagonists have been unsuccessful, although several inhibitors of the mitogen-activated protein kinase (MAPK) p38, which is part of the signaling cascade activated when TNFα binds to its receptor, are still in development (7). Efforts were thus focused on technologies that had only recently become commonplace in the research lab: 1) monoclonal antibodies (MAbs), and 2) recombinant DNA cloning and expression. MAbs were ideal due to their high solubility, exquisite specificity and long serum half-life. Recombinant DNA techniques have made protein engineering routine, as virtually any protein can be modified to provide the optimal qualities required. The evolution of these technologies continues to drive the development of biotherapeutic agents.
Monoclonal Antibody Technology
The technology to generate and select cell lines that would produce a single specific antibody was reported first by Kohler and Milstein in 1975 (8) . These cell lines are produced by the fusion of splenocytes (which make antibodies) harvested from immunized mice with an immortalized murine myeloma cell line (Figure 1⇓). The surviving fused cells, or hybridomas, continue to secrete the same antibody as the parent splenocyte and retain the immortal qualities of the parent myeloma cell. Each hybridoma synthesizes one “monoclonal” antibody that binds to a specific region (i.e., epitope) on the antigen used to immunize the mouse. The antibodies secreted by each of the hundreds of individual hybridomas normally produced from the fusion of a single mouse spleen are then screened for specificity and affinity of binding to the antigen. The most potent antibodies are then screened for functions mediated by the Fc portion of the antibody molecule (Figure 1⇓) such as complement fixation.

Standard method used to generate murine MAbs. Splenocytes from immunized mice are fused with mouse myeloma cells to generate antibody-secreting, immortalized hybridoma cells. The monoclonal antibody produced by individual hybridoma cell clones are then screened for optimal antigen binding, as defined by the complimentarity-determining regions (CDR) on the Fab portion of the antibody, as well as for the desired isotype, as defined by the Fc portion of the antibody.
In 1986, just eleven years after Kohler and Milstein’s original publication, a murine MAb (muromonab-CD3) that recognized the CD3 antigen found on human T cells was approved by the Food and Drug Administration for the treatment of kidney transplant rejection. Patients were limited to a single 10- to 14-day dose regimen with this product because the majority of patients developed a significant immune response during treatment (9) . The immunogenicity of murine MAbs in human patients, as well as the fact that many hybridoma cell lines were not very stable, threatened to severely limit the usefulness of this technology. However, further developments in protein engineering techniques would provide the tools needed to overcome these issues.
Recombinant DNA Technology Improves MAbs
A straightforward and simple improvement applied to MAb technology was the generation of a chimeric antibody, in which the murine constant domains of the desired murine MAb are replaced with human constant domains (Figure 2⇓). This strategic step, alleviating the possibility of a human patient developing an immune response to the murine constant domains on the mouse MAb, was utilized in the production of the chimeric TNFα -specific MAb infliximab (Figure 3⇓). The binding characteristics observed with the fully murine antibody are identical to those of the chimeric antibody (10) , and the human fragment of the MAb consisting of the constant domains (i.e., Fc portion) of the chimera (a human IgG1 isotype) was still functional (11) . For an antibody to be complete it must be properly glycosylated; therefore, antibodies must be produced in mammalian cell lines. In the case of infliximab, a murine myeloma cell line was used. Human studies comparing murine and chimeric antibodies have demonstrated greatly reduced immunogenicity with a chimeric antibody (12).
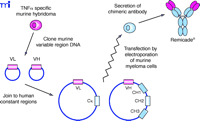
Derivation of chimeric mouse–human MAbs such as Remicade® using recombinant DNA technology. Heavy and light chain variable cDNAs are cloned from a hybridoma cell clone producing the desired mouse MAb . These variable sequences (VL and VH) are inserted into plasmid vectors containing the desired human light and heavy chain constant domain sequences (the Cκ and G1 CH1, CH2 and CH3, respectively) and transfected into myeloma cells. The antibody secreted by these cells is now chimeric, containing variable protein domain derived from the original mouse antibody fused to human constant protein domains.
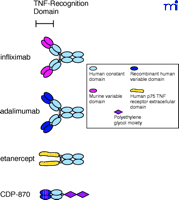
Schematic drawings of the four TNFα antagonist biologics now available or in clinical development. Structurally, infliximab and adalimumab are similar to normal human immunoglobulins. Etanercept contains the TNF receptor extracellular domain fused to the heavy chain CH2 and CH3 constant domains of human immunoglobulin. CDP-870 includes only the Fab component of a human antibody that is further modified by the attachment of a polyethylene glycol moiety.
A more complicated procedure, referred to as humanization, requires the replacement of the murine framework sequences around each complementarity-determining region (CDR), which recognizes and binds to the antigen’s epitope, with human framework sequences, in addition to the replacement of the constant domains (Figure 4⇓). CDP-870 was produced using this strategy (Figure 3⇑), and represents another approach to inhibition of TNFα ; removal of the Fc portion of the antibody eliminates any potential Fc-mediated functions. The safety and efficacy of CDP-870 are currently being tested in Phase II and Phase III clinical trials in both RA and Crohn’s disease patients. Because the framework holds the CDRs in the proper orientation, it is not unusual to see a decrease in antigen-binding affinity when murine antibodies are humanized in this manner, and additional mutations in the CDR sequences may be necessary to optimize affinity for the antigen. CDP-870 incorporates only the fragment of an antibody consisting of the variable A and B sequences responsible for binding the epitope, termed the Fab portion, which is typically not glycosylated; therefore, CDP-870 can be expressed in bacteria. The smaller Fab portion of the antibody typically has a much shorter serum half-life, and hence the molecule is further modified by the addition of a polyethylene glycol (PEG) moiety (Figure 4⇓), which reportedly increases the half-life and reduces the immunogenicity of the protein (13).
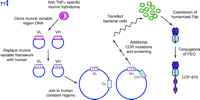
Derivation of CDP870 from a murine TNFα -specific MAb using recombinant DNA technology and protein engineering. The cloned murine variable light and heavy chain cDNAs are modified to insert human framework sequences, and thus only the CDR sequences remain from the original mouse monoclonal antibody. Because this may reduce affinity for the original antigen, additional site-specific mutations may be necessary within the CDR sequences. Because the native Fab is typicall not glycosylated, CDP-870 can be expressed in bocterial cells; PEG is conjugted to the purified humanized molecule.
Recombinant DNA techniques have also been used to produce a different type of TNFα antagonist comprising the extracellular domain from the human TNF receptor, expressed as a fusion protein with the Fc portion of a human antibody (Figure 5⇓). The two known TNF receptors are p55 and p75, and etanercept was designed using the extracellular domain of the p75 receptor. The use of the receptor itself to bind TNFα lends specificity, although TNF receptors are known to bind both TNFα and a related cytokine called lymphotoxin (or TNFβ ). The antibody-derived Fc portion of the molecule increases the serum half-life and, because it is dimeric, provides two TNFα binding sites per molecule. Despite the derivation of etanercept sequences from human sources, immunogenicity has been reported against the unnatural protein sequence generated by the fusion of the two proteins. Interestingly, a similar fusion protein that utilized the extracellular domain of the p55 receptor (lenercept) failed in RA clinical trials primarily due to a high degree of immunogenicity (14). This scenario illustrates the many factors, including glycosylation, aggregation, route of administration, dose regimen, and formulation (15), that may contribute to immunogenicity, even in the case of a totally human protein sequence.
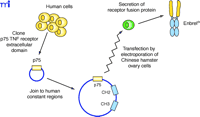
Generation of the receptor–immunoglobulin fusion protein Enbrel®. Sequences coding for the p75 TNF receptor extracellular domain, cloned from a human cDNA library, are inserted into a plasmid vector containing the CH2 and CH3 heavy chain constant domain sequences and subsequently transfected into Chinese hamster ovary cells. These cells secrete a homodimeric fusion protein because of the interchain disulfide links between the two CH2 domains.
Human MAbs, such as adalimumab (Figure 3⇑), have also been derived from a murine MAb template using a technique called phage display (Figure 6⇓). Antibody fragments can be displayed on the surface of phage particles as fusion proteins (16). Using the TNFα -specific murine antibody as a template, two phage libraries were established, one with the TNF-specific murine light chain paired at random with human heavy chains, and the second with the TNF-specific murine heavy chain paired at random with human light chains. Human heavy and light chains from the two hybrid phage libraries that still bind to TNFα were isolated, paired together and selected again for binding to TNFα . As with humanization, such as process can result in a human antibody with reduced affinity for the antigen; hence, additional mutations in the CDRs are often necessary. The phage-derived variable domains were fused with human constant domains and inserted into an appropriate mammalian cell line––Chinese hamster ovary cells in the case of adalimumab––for production. Although all its framework and constant domain sequences are human-derived, adalimumab still elicits an immune response in about 5% of RA patients; the immune response is apparently directed to the CDRs and reduces clinical efficacy. This result is not surprising, as the hypervariable CDRs for any antibody are by definition a unique sequence that can generate an immune response (15).
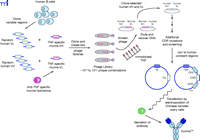
Generation of a recombinant human antibody (Humira™) from a murine TNFα -specific MAb and human variable gene phage display libraries. Two phage libraries containing the TNF-specific VL and VH sequences from the murine hybridoma cells paired with random VH and VL sequences derived from human B-cells are screened for binding to TNFα. The paired human VH and VL sequences from positive clones express phage-displayed human Fab molecules that bind to TNFα but require additional site-specific mutations in the CDRs to increase binding affinity to an acceptable level.
Success of Bioengineered Proteins in the Clinic
As described above, three bioengineered MAbs and one TNF receptor fusion protein have been developed as TNFα antagonists. Key differences in the molecular characteristics of these TNFα antagonists are shown in Table 1⇓. Three members (infliximab, etanercept and adalimumab) of this new class of biotherapeutic are currently licensed for treatment of RA and are described below. The fourth (CDP-870) has shown promising results in phase II RA trials; phase III clinical trials are underway.
Anti-TNFα Biotherapeutics Currently Licensed for the Treatment of RA
Infliximab (Remicade® , Centocor, Inc) binds to all forms of TNFα (soluble monomer, soluble trimer, transmembrane, and receptor-bound; 17 ) and was safe and effective in a two-year pivotal phase III clinical trial termed the ATTRACT (a nti-T NF t herapy of r heumatoid a rthritis with c oncomitant t herapy) trial. Infliximab can mediate lysis of cultured cells expressing transmembrane TNFα . Following an induction regimen of 3mg/kg administered by intravenous infusion at zero, two, and six weeks, efficacy can be maintained with additional infusions every eight weeks. If necessary, dosing can be adjusted incrementally up to 10 mg/kg, or the dose frequency can be reduced to every four weeks. Infliximab has a serum half-life ranging between eight and ten days (10) , and antibodies to infliximab were detected in 8.5% of the infliximab-treated patients. Based on results of the ATTRACT trial, infliximab, in combination with methotrexate, has gained approval for reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in patients with moderately to severely active RA (18).
The receptor fusion protein etanercept (Enbrel® , Immunex Corporation) binds to the soluble and transmembrane forms of both TNFα and lymphotoxin (17). It is administered subcutaneously at a dose of 25 mg twice weekly and was demonstrated to be safe and effective in a series of three pivotal phase III clinical trials that remained blinded for six or twelve months. The serum half-life for etanercept is 4.8 days, and antibodies to etanercept can be detected in about 5% of etanercept-treated patients. Etanercept is indicated for reducing signs and symptoms and inhibiting progression of structural damage in patients with moderately to severely active RA and may be given alone or in combination with methotrexate (19).
The recombinant human MAb adalimumab (Humira® , Abbott Laboratories) binds specifically to TNFα and is capable of lysing cells expressing cell surface TNFα in vitro in the presence of complement. Efficacy and safety were demonstrated in a series of pivotal phase III clinical trials over the course of six to twelve months that tested adalimumab alone (monotherapy) and in combination with methotrexate. The terminal half-life in serum ranged from fourteen to nineteen days, and the recommended dose is 40 mg administered subcutaneously every other week. Weekly doses of 40 mg are permitted, but only in the absence of methotrexate. Approximately 5% of the treated patients developed antibodies to adalimumab and patients who developed an immune response had reduced efficacy. Adalimumab is indicated for reducing signs and symptoms and inhibiting the progression of structural damage in adult patients with moderately to severely active RA, either alone or in combination with methotrexate (20).
Successful Treatment of Psoriasis With a Bioengineered Protein
Psoriasis is a chronic, inflammatory disease in which T cells infiltrating the skin are activated and subsequently proliferate and secrete proinflammatory cytokines such as TNFα and interferon–γ . Although seldom life-threatening, this disfiguring disease can have a significant psychological effect on patients with moderate-to-severe psoriasis (21). Immunosuppressive treatments such as methotrexate, cyclosporin, and phototherapy (psoralen and ultraviolet A), all known to broadly target T cells, led to the development of a more specific T cell biologic called alefacept (Amevive® , Biogen). Alefacept is a dimeric fusion protein that consists of the first extracellular domain of the human leukocyte function antigen-3 (LFA-3) linked to the Fc (hinge, CH2 and CH3 domains) portion of human IgG1. LFA-3, normally found on antigen-presenting cells, binds to the cell surface protein CD2 found on memory T cells, which allows the T cells to become activated. The LFA-3 portion of alefacept that binds to CD2 therefore blocks the co-stimulatory signal required for T cell activation. In addition, it is hypothesized that the Fc portion of alefacept could bind to the Fcγ RIII receptor found on NK cells and macrophages, thereby inducing apoptosis of the T-cell population driving the disease process.
Alefacept is similar in structure to etanercept, and is produced by recombinant DNA technology in a Chinese hamster ovary expression system similar to etanercept and adalimumab. As for most antibodies and Fc fusion proteins, alefacept has an extended serum half-life (12 days), and the volume of distribution in patients (94 mL/kg) suggests that it occupies primarily the vascular space (22). Although all the sequences are derived from human genes, about 3% of treated patients develop an immune response to alefacept (23).
In clinical trials, alefacept treatment, compared with placebo, was associated with dose-related significant improvements in the Psoriasis Area and Severity Index (PASI) score from baseline. The recommended dose regimen for alefacept is 7.5 mg given once weekly as an intravenous bolus or 15 mg given once weekly as an intramuscular injection for twelve weeks, which may be repeated provided that CD4+ T lymphocyte counts are within the normal range. Alefacept is approved in the United States for the treatment of adult patients with moderate-to-severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy (23).
Conclusions
New biologics have resulted from steady scientific progress and technological breakthroughs that now provide effective new tools for protein engineering. The theoretical role of TNFα -specific agents in patients with RA has been confirmed in a number of clinical trials with each of these agents. To date, hundreds of thousands of RA patients in the US and Europe have been treated with an engineered protein, infliximab or etanercept; adalimumab has also recently been approved for the treatment of RA.
Although lessons learned and hypotheses tested have led to the relatively swift and safe implementation of these biologics, not all “designer proteins” have worked as expected. These failures have taught us that we may not know all the rules concerning how the human body will react to an engineered protein, and that each new therapeutic must go through rigorous clinical testing and careful monitoring once in full commercial use. Despite these trials and errors, the vast majority of RA patients who have received a TNFα antagonist have experienced minimal side effects and significant efficacy not seen prior to the use of these agents.
The introduction of these new of TNFα -specific agents has added a new dimension to the treatment of RA based on their ability to inhibit permanent structural damage and reduce disability. In addition, these technologies have already been used to develop a whole new generation of biologics that have been shown to be safe and effective in a variety of other chronic diseases such as asthma, organ transplantation, and cancer.
Acknowledgments
Centocor, Inc provided support for the preparation of this manuscript.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Roy Fleischmann, MD (top), is a clinical professor at the University of Texas Southwestern Medical Center. Dave Shealy, PhD (bottom), is Associate Director of Protein Biochemistry at Centocor, Inc. Address correspondence to RF. E-mail royfleischmann{at}radiantresearch.com; fax 214-879-8877.

