Sir James Black: Learning by Doing
Talking to Sir James Black, it is hard to understand why anyone would choose a career other than biomedical research. It’s not that he advocates the profession explicitly—in fact, he does not have much patience for those who would scrutinize his accomplishments and motivations in an effort to extract lessons for success. Rather, his historical perspectives have a way of making the elucidation of key concepts highly accessible —even poetic—and of making unanswered questions seem important and alluring. His head is full of dates and names from history, but it is a passion for the scientific method that permeates his speaking and writing. He entered the pharmaceutical industry (ICI), at a time when such a move was regarded somewhat skeptically, to pursue his idea of developing drugs to block the effects of adrenaline on the heart affected by coronary artery disease—a concept that was regarded very skeptically. And after the spectacular success of bringing beta-blockers into clinical use—indeed, to avoid the spectacle of success—he moved to a second company (Smith, Kline & French), where his seminal research in gastric acid secretion led him to develop cimetidine. These achievements and his subsequent work, both in academia and in industry, have identified Sir James as a landmark figure in biomedicine from our own time. In 1988, along with Gertrude Elion and George Hitchings, he was awarded the Nobel Prize for his work in drug discovery.

MI: What kind of career were you planning when you first went off to St. Andrews?
JB: I have lived an unplanned life. But for an accident, I wouldn’t have gone to university—my parents couldn’t have afforded it. I was fourth in a family of five boys. A math teacher persuaded me to sit for the entrance exams, and although I didn’t feel hopeful, I got offered a residential scholarship.
MI: And once you got to university, did you know what academic direction you wanted to take?
JB: Studying medicine was an option that had become apparent because of my older brother, who had been bringing home his textbooks during his medical training—his physiology books particularly excited me.
MI: Did you think you would go into clinical practice?
JB: I think I could have enjoyed doctoring, actually. I never found any problems dealing with personal intimacy, but I couldn’t take the way that doctors treated patients at that time. The patients were sort of dehumanized. I found the science of medicine interesting because of the breadth of it. And in those days we had to do basic subjects like chemistry, physics, and zoology. The professor of zoology was D’Arcy Thompson, and he must have been eighty years old then, I suppose. He was a Darwin look-alike, I mean, big Fedora black hat, black cloak, parrot on his shoulder, and of course his book On Growth and Form—it’s just a joy to read.
MI: So, you were too nice a person to become a doctor under those conditions!
JB: I don’t know if I’m a nice person…I don’t even know if I’m a scientist!
MI: Really??
JB: Well, I’m untrained, you see. I’ve never had any formal instruction in experimental science—I’ve just picked it up. I haven’t done a PhD.
MI: So, where did you “pick it up”?
JB: After completing medical school, I went round and knocked on my professor’s door, and I got involved in Professor Robert Garry’s laboratory—they were doing work on the intestinal absorption of sugars. Professor Garry was having a head-to-head with Verzar, a Swiss physiologist, who believed that the selective absorption of sugars had to do with phosphorylation—he was using sodium iodoacetate to poison phosphorylating enzymes. Garry’s research assistant showed that during the time course of Verzar’s observations, the treatment with iodoacetate was completely destroying the epithelium—hence, no “selective” absorption. And so I remember asking Garry’s assistant, “What does this poison do?” And her hands went up. I asked, “What happens to blood pressure?” She didn’t know, and so I made a little narrow- bore sphygmomanometer, and I showed that the rats that they were working with underwent a great loss of blood pressure. The question for me then was, you’ve done something to the epithelium to interfere with sugar absorption, but you’ve also done something very bad to the blood pressure: Does blood pressure influence absorption of sugars? It was then that I developed this parallel interest in cardiovascular physiology and gut physiology. That year of research really mapped out my life.
MI: After that year of experimental research in Dundee, you decided to leave. Why?
JB: Well, it was a year in which I got married, and my scholarship was only for four years, so I had acquired what seemed to me massive debts. And so I did what any Scots boy would have done—I went elsewhere. I found a lectureship in Singapore. Of course, for me, it was also a matter of doing my research. I had to make my own equipment—I made low-volume elasticity displacement manometers out of tubes covered with very thick rubber on which I glued a concave mirror, which reflected the light from a filament I had set up in a dark room. I had set up a big box containing a glass rod that would focus the reflected light and give a displacement measurement. I had to work at night anyway, because it was so hot. When you work like that, you become intellectually and emotionally isolated. You don’t know whether your research is great or absolute rubbish. You’ve no way of judging yourself!
MI: Well, how did you see yourself at that point and the direction you wanted to take?
JB: When you ask questions like that, all I can say is, “I don’t know!” I was nearly disappearing into outer space. I suppose I was intuitively following a reductionist path, and I was getting more and more down to the mathematics and physics of rheology and so on. It’s not that you plan to go that way—you’re not programmed. When students ask me, “What should I be doing?” I can only respond, “I don’t know! What have you been doing up to now?”
MI: So, what did you do when your three years in Singapore ended in 1950?
JB: I went around to physiology departments in London knocking on doors. One day, feeling a bit low, I was walking in London and I felt a hand on my shoulder—it was Professor Garry. He directed me to a chap called Bill Weipers, who had just been made Director of the Glasgow Veterinary College, created by an act of Parliament in 1949. Up to that time, there had been no academic veterinary departments in the U.K., but Weipers was the best-known small-animal veterinarian in the west of Scotland. He didn’t have the academic credentials to be appointed Professor, which is why they made him Director! I went to meet him for an interview, and I just found him magnetic. So, here I was, still wet behind the ears, untrained, and given the opportunity to build up a physiology department from absolutely nothing. I was lucky, because I could build up my own practical classes, lectures, and research. There was a small company in London at the time getting government support to develop domestic technologies, including recorders for cardiologists—ECG recorders and manometers. I started working with them, and as a result, our funny old building in Glasgow was outfitted with advanced equipment that wasn’t available anywhere else in the university. So, I was able to recruit two surgeons in Glasgow—Adam Smith and George Smith.
Adam Smith had worked on 5-HT, which was hot news in the late 40s, and he had shown that it didn’t stimulate gastric acid secretion, but increased the amount of mucus. We started doing experiments on gastric secretion, looking at the interaction with 5-HT and histamine. George Smith was a cardiovascular surgeon who had been trained in Baltimore, and he’s the one who introduced hyperbaric oxygen by first showing that it could stabilize fibrillating dog hearts. I knew just enough chemistry to know that oxygen has a very low solubility in water, and that at a fifth of an atmosphere, blood hemoglobin is fully saturated. So, two atmospheres can only be increasing the dissolved oxygen by only a few percent. I began to think at that point, that instead of increasing oxygen supply, why don’t I decrease the demand for oxygen by the heart?
MI: So, you were thinking along these lines quite early on.
JB: Oh yes. Once you get an idea, it takes a long time to evolve. I knew just enough pharmacology to know that sympathetic drive determines heart rate, so why not reduce that drive by using anti-adrenaline drugs in the heart. And of course, once you investigate this, you find that when you’re lying down they don’t do much; when you stand up, your blood pressure falls, but your heart rate goes up. Henry Dale had shown this (1906) with his own dogs when he gave them ergot. So, it was obvious why no one had ever looked at the anti-adrenaline drugs in angina, because the one thing you don’t want to do is lower the core pressure.
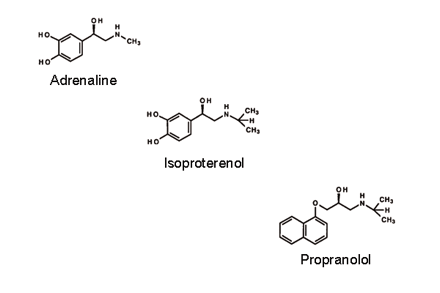
Now, as an accident, I had bought Drill’s Pharmacology in Medicine—a lovely book, beautiful paper—and there is this discussion by Raymond P. Ahlquist in which he proposed the existence of a dual (α and β) adrenoceptor system. Adrenaline activated α receptors to constrict blood vessels and raise blood pressure; adrenaline activated β receptors to increase heart rate and force. Now, I never managed to find out why Victor Drill had asked Ray Ahlquist to write these two chapters, the inclusion of which is surprising, because at that time his proposal had been published only with great difficulty—it just wasn’t in the medical dogma. I just came across it—I was just trying to teach myself some pharmacology —trying to teach myself something about physiology! Well, at that point I see what I want to do; we have to get a drug that will block the actions of adrenaline on the β-receptors.
MI: Can you explain why Ahlquist’s proposal of multiple receptor types encountered such resistance?
JB: Oh yes, I think I can answer that. The word receptor had been around since the end of the nineteenth century, but physiologists had used the word receptor only generally: pressure receptors, sensory receptors, chemoreceptors, etc. Among the endocrinologists, Starling introduced the term hormone in 1905, laying the foundation for endocrinology as a form of communication, but endocrinologists—the people who were going around extracting glands and talking endlessly about the endocrines—didn’t in fact talk about how hormones act. The only people who talked about receptors as interactive entities was a small cadre of pharmacologists who were interested in the quantitative relationship between dose and response, because to apply the law of mass action you had to have a drug acting on something. For these people, the word “receptor” was a pure idea, necessary to allow them to do the math. But they were embarrassed about it, that it was a pure invention, which is why Cannon and Rosenblueth proposed multiple forms of adrenaline, referring to alternative forms as “sympathin E and sympathin I.” And so when Ahlquist claims to find two receptor types—that stuck in everybody’s throat. His was the first explanatory use of the term “receptor.”
MI: So, you had the idea to develop a beta-blocker. How do you go about creating a drug from an idea?
JB: One thing was clear at that time: going into industry was a no-no. If you were a good scientist, you didn’t go into industry. However, I had this friend who was a pharmaceutical rep for ICI, and it was he who persuaded me to contact them. ICI had built this wonderful new lab down in Cheshire, and I approached them to see if I could get some money from them for my research. I wrote to them, laid out my story, and I got a site visit! Two guys visited and expressed their interest, but they thought it would be easier if I would go work with them at the new campus. I went to see it, and the place was magnificent—what a contrast from the old place in Glasgow. I was given a chemist, John Stephenson, to work with, and off we went!!
MI: Did you know what to do with a chemist?
JB: Yes. Because we knew, simply, that the defining molecule for β-receptors was isoproterenol. So, we wrote down the structure, compared it to noradrenaline, a primary amine, and to adrenaline with its methyl group; with those two natural ligands, you bind to a different mix of α- and β-receptors. When you increase the size of the substituent on the amine nitrogen a wee bit by changing the methyl group to an isopropyl group (in isoproterenol), you lose activity at α-receptors. That’s all we knew. So, we thought that if we increased the substituent size further, maybe we would get things that begin to block specifically. We made no progress until Powell and Slater described the properties of DCI, a ring-substituted derivative of isoprenaline that we now recognize as a potent partial agonist. We thought that by concentrating on ring substitution we might find something devoid of agonist activity, which led us to propranolol.
MI: The medicinal chemistry approach that you described in developing propranolol—that rather set the pace for a new era of pharmaceutical development, didn’t it?
JB: Well, every successful medicinal chemist that I’ve known has started with a concept and then thought up the techniques necessary. Paul Janssen, for example, our most fecund drug discoverer or inventor ever, always started that way. His first idea began with pethidine (meperidine), introduced as an antispasmodic around 1940 and subsequently recognized as an addictive analgesic. Paul knew that pethidine was a piperidine derivative, a simple molecule with simple chemistry, and he had the idea to try to separate the drug’s two properties. He eventually produced loperarnide, an antispasmodic, and fentanyl, an analgesic. If you look at their structures, they’re all like a family tree. Paul, like most medicinal chemists in the 50s, started from compounds known to be active in man. When we started looking for beta-blockers, we did not have the luxury of a known compound. All we had was isoproterenol, a selective agonist. When we started the H2-receptor project, we began to focus on the effects of 4-methylhistamine, a selective H2-receptor agonist. Later on, I deliberately started from the messenger molecule; we did this with 5-HT and gastrin. This approach, of turning agonists into highly selective antagonists seems to work well, but is invariably slow. All of us who did these sorts of things—it really is a sort of forced Darwinian evolution: we as medicinal chemists would force “mutations” in a molecule, and then just as in Darwinian evolution, you expose the new entity to an environment, asking, “Is its survival better according to our bioassay?”
MI: Do you think there’s now an overemphasis on technique that distracts people from doing the job of “evolving drugs” by rational discovery?
JB: Well, I worry about it. Nowadays, we’re obsessed with techniques, whether it’s techniques of accumulating information in databases, or developing chemical models, it’s all technique, technique, technique. With the development of combinatorial chemistry and high-throughput screening, you have to buy molecules by the 100,000s because your assay system will gobble them up. But these molecules are thrown together by reactions tailored for high yield in moderate conditions, and the molecules that come out in the end are highly dependent on the three bond angles of carbon —so you get “leads” that are not biological, biochemical look-alikes. In the old-fashioned iterative method that I’ve talked about, we always started with a biological lead. I have never ever started without such a lead! I’ve had isoproterenol; I’ve had histamine; I’ve had tetragastrin. Because it’s an iterative process, it is slow. But what’s the hurry? A big drug company cannot develop fully more than about one drug at a time, due to the huge technological business of getting the trials done, getting the message out, teaching the people in the organization the technology behind it—it’s a huge learning curve.
MI: But that isn’t the story that one hears about modern pharma!
JB: But modern pharma is not delivering. The expenditure of R&D is going up, and output of new drugs is falling, which is big trouble. Now, mind you, this is just my prejudice. But there’s so much effort going in the direction of technology, and so much money, and people are being forced to make promises. Well, impatience is not a bad thing, unless it so happens that the technology to satisfy impatience only appears to have been developed. In the old days, we didn’t have to make promises about our research—I never promised anybody anything in my life. I mean, I never even promised to work hard!
- © American Society for Pharmacology and Experimental Theraputics 2004

