Ubiquitination for Activation: New Directions in the NF-κB Roadmap
Interactions between antigens and T cell receptors (TCRs) stimulate several signaling cascades that activate numerous transcription factors including nuclear factor of activated T cells (NFAT), Activator protein–1 (AP-1), Ets, and nuclear factor–kappa B (NF-κB). The activity of the NF-κB family is also induced by other factors such as cytokines or tumor necrosis factor (TNF) family members, making NF-κB one of the most studied transcription factors in the immune system. The mammalian NF-κB family consists of many members including Rel-A (p65), NF-κB1 [p50 (mature), p105 (precursor)], NF-κB2 [p52 (mature), p100 (precursor)], c-Rel, and Rel-B, all of which are regulators of B and T lymphocyte functions. NF-κB exists in the cytoplasm in an inactive form associated with the IκB (inhibitor of NF-κB) proteins, which include IκBα, IκBβ, and IκBε. These molecules contain small ankyrin repeats, which mediate protein–protein interactions. In addition, the NF-κB precursor proteins p100 and p105 contain similar ankyrin repeats in their C termini and, thus, can also function as IκB-like proteins (1, 2).
The binding of NF-κB to IκB results in the masking of nuclear localization sequences (NLS) on NF-κB. Upon T cell stimulation, in response to many different mediators such as TNFα, IL-1β, or direct stimulation of CD3 (the major signaling chain in the TCR complex) and CD28 (a costimulatory protein located near the TCR whose signals are required for NF-κB activation), cascading pathways are switched on, resulting in the activation of IκB kinases (IKK). The IκB kinase (IKK) signalosome, which possesses two nearly identical subunits (IKK-α and IKK-β) and a regulatory subunit [IKK-γ or NF-κB essential modulator (NEMO)], phosphorylates two regulatory serines located in the N terminus of IκB, triggering ubiquitination and subsequent proteolysis of IκB in the 26S proteasome complex. Degradation of IκB molecules unmasks the NLS of NF-κB, allowing it to rapidly translocate into the nucleus where it regulates the transcription of a wide variety of immune responsive genes.
Very recently, the characterization of three additional proteins––Bcl10 (B cell lymphoma 10); CARMA1 [Caspase recruitment domain (CARD), membrane-associated guanylate kinase (MAGUK) protein 1]; and Malt1 (Mucosal-associated lymphoid tissue lymphoma translocation protein-1)––has allowed for the development of a feasible paradigm for the TCR-induced activation of NF-κB.
Bcl10 (also known as CLAP/CIPER/cE10/CARMEN), originally identified from the recurrent chromosomal translocation breakpoint t(1;14)(q22;q32) in B cell lymphomas of the mucosaassociated lymphoid tissue (Malt) type (3), is an adaptor protein containing an N-terminal CARD (Figure 1A⇓). Recent studies by Mak and colleagues, using Bcl10−/− mice, demonstrated that Bcl10 functions as a fundamental regulator of both adaptive and innate T and B cell responses (4). Bcl10 had been previously earmarked as a proapoptotic molecule; however, Bcl10−/− mice have no apparent apoptotic dysfunction, but rather mice exhibit a dramatic reduction in all serum immunoglobulin (i.e., antibody, Ig) isotypes and are severely impaired B and T cell responses to infections in vivo. The further observation that Bcl10−/− T and B cells lacked the ability to activate NF-κB by either B cell receptor or TCR–CD28 indicated that Bcl10 is a crucial participant in the regulation of NF-κB activation.
CARMA1 (also known as CARD11 and BIMP3) was originally identified through searches of protein databases (5, 6). CARMA1 possesses a coiled-coil domain, a PDZ [postsynaptic density 95 (PSD-95), discs large (Dlg), and zonula occludens-1 (ZO-1)] domain, an SH3 (Src homology 3) domain and a C-terminal GUK domain, and is expressed mainly in lymphoid tissues. CARMA1 binds directly to Bcl10 through homophilic CARD–CARD interactions. The overexpression of CARMA in Bcl10-deficient cells cannot overcome the inability to activate NF-κB, thereby placing CARMA1 “upstream” of Bcl10 in the pathway of TCR-mediated NF-κB activation (7). CARMA1-deficient T-cell lines have defective TCR-mediated NF-κB activation. Moreover, CARMA1 was placed upstream of protein kinase C–?? (PKC??), a key signaling molecule in CD3–CD28 costimulation, and is thought to provide a functional link between PKC??activation and Bcl10 (5, 8, 11). CARMA1 knockout mice exhibit defective B-1 and NK cell development and defective B- and T-cell responses (12, 13). Several groups have simultaneously identified a role for CARMA1 and Bcl10 in NF-κB activation. However, neither molecule can bind directly to IKK-γ/NEMO, suggesting that further intermediates exist. Enter Malt1.
Malt1 was originally identified from a recurrent chromosomal translocation in mucosal–associated lymphoid tissue lymphomas (14–16). The t(11;18) breakpoint fuses the C terminus of the Malt1 protein to that of the N terminus of c-IAP2 (inhibitor of apoptosis protein 2, also known as API2). Malt1 contains an N-terminal death domain, two Ig-like repeats, and a C-terminal region that resembles the catalytic domain of caspases and, therefore, has been referred to as a mammalian paracaspase (17). Evidence suggests that Bcl10 can use Malt1’s two Ig-like repeats to induce oligomerization of Malt1. Furthermore, coexpression of Bcl10 and Malt1 demonstrate enhanced Bcl10-mediated NF-κB activation, suggesting that Malt1 may be involved in the regulation of IKK activity (18). In addition, c-IAP2–Malt1 fusion proteins are strong activators of NF-κB alone (17, 18).
More recently, the Dixit and Mak groups have shown, that Malt1 is crucial for TCR-induced NF-κB activation in B and T cells (19, 20). Both groups observed that Malt1-deficient mice have decreased numbers of B cells found in the margin of the spleen, alterations in early thymocyte subpopulations, and fewer peripheral activated T cells. Malt1−/− mice failed to produce IgM and IgG upon immunization, and amounts of all Ig isotypes were reduced, suggesting a critical role for Malt1 in the adaptive immune response. Strikingly, NF-κB from Malt1-deficient T cells is unable to bind nuclear DNA; however, this was observed to be a TCR-specific phenomenon because stimulation with TNF-α or IL-1 resulted in normal NF-κB activation. Malt1 does not require Bcl10 for NF-κB activation and acts downstream of Bcl10, but Malt1 is upstream of IKK because catalytic IKK activity could not be induced in either Malt1−/− or Bcl10−/− T cells (20). Notably, wild-type and Malt1−/− T and B cels exhibited comparable antigen receptor—proximal signaling and mitogen-activated protein kinase (MAPK) activation.
So where exactly does Malt1 act in TCR-activated signaling? To answer this question the Lin and the Dixit group joined forces and demonstrated that Malt1 is recruited into lipid rafts at immunological synapses following TCR–CD28 stimulation (21). Using a yeast two-hybrid system, they showed that Malt1 interacts directly with the coiled-coil domain of CARMA1 via its caspase-like domain. CARMA1 was also critical for the recruitment of Malt1 to lipid rafts because CD3–CD28 costimulation fails to recruit Malt1 into lipid rafts in CARMA1-deficient T cells. They also observed that Malt1 not only binds to Bcl10 directly but also associates with CARMA1 in a Bcl10-independent manner, suggesting that Malt1, Bcl10, and CARMA1 form a trimolecular complex. Intriguingly, a Malt1 mutant containing only the N-terminal death domain and the Ig-like domains inhibited TCR-induced activation of NF-κB, whereas a Malt1 mutant containing the caspase-like domain enhanced this response. Therefore, Malt1 can act as both a negative and positive regulator of TCR-induced NF-κB.
So, how do these molecules interact with the IKK signalosome in order to allow activation of NF-κB? Remarkably, Dixit and his colleagues have shown that Bcl10 targets NEMO. By using overexpression and small inhibitory RNA (siRNA) techniques, they demonstrated that Malt1 and the ubiquitin-conjugating enzyme UBC13 are both required for Bcl10-induced NEMO ubiquitination and subsequent NF-κB activation. It has been proposed that Bcl10 activates Malt1 by inducing a conformational change and subsequent Malt1 oligomerization. The ubiquitin-conjugating E2 complex consisting of UBC13 and MMS2 binds to Malt1 (Figure 1B⇓) and catalyzes formation of a polyubiquitin chain at K63 on NEMO, leading to the destruction of NEMO through the 26S proteasome. NEMO’s removal might then effect the recruitment of an upstream regulator, such as a putative IKK kinase, to activate the IKKs or may induce a conformational change within the IKKα–IKKβ complex that leads to autophosphorylation and activation (22).
In addition to NEMO, Bcl10 itself is the target of the nearly identical ubiquitin ligases NEDD4 and Itch. These two molecules promote ubiquination of Bcl10 upon CD3–CD28 stimulation, resulting in attenuated NF-κB activation (23). Thus, a feedback mechanism exists in which Bcl10-mediated NEMO degradation drives initial NF-κB activation but later its degradation prevents excessive signaling (Figure 1B⇓).
Linking TCR-synapse engagement to NF-κB activation has been the subject of extensive research in the past; however, recent discoveries have added substantially to our understanding of this pathway. Ubiquitination of these molecules also regulates signaling cascades. Importantly, these findings suggest that unique selective regulation of the broadly utilized transcription factor NF-κB does occurs downstream of TCR activation. Considering the involvement of several of these new molecules in immune-related diseases and several types of cancer, these findings will surely give new impetus to future research in this area to exploit these and other still unknown targets for the treatment of immune diseases.
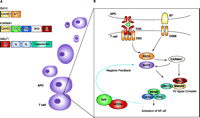
Mapping NF-κB activation in T cells. A. Domain structure of Bcl10, CARMA1, and MALT1. Domains within each protein may promote homo-oligomerization and multiprotein complex formation. CARD, caspase recruitment domain; S/T, serine-threonine-rich domain; CC, coiled-coil domain; PDZ, postsynaptic density 95, discs large, and zona occludens-1 domain; SH3, Src homology 3 domain; GUK, guanylate kinase domain; DD, death domain; Ig, immunoglobulinlike repeats; caspase-like, a domain very similar to those found in caspase proteins. B. Upon TCR activation, CARMA1 interacts with Bcl10 at the immunological synapse via their CARD domains. CARMA1 also recruits PCK??to the lipid rafts at the synapse, which may further aid Bcl10 activation. Through its coiled-coil domain, Malt1 binds CARMA1. Malt1 is, itself, recruited to the rafts. Bcl10 induces the oligomerization of Malt1, allowing for the formation of a ubiquitin-conjugating E2 complex consisting of UBC13 and MMS2. These molecules catalyze K63-linked polyubiquitination of NEMO (leading to its destruction) and the activation of IKK. Ubiquitin ligases NEDD4 and Itch can also negatively regulate this pathway by degrading Bcl10.
Acknowledgments
The authors wish to thank Jim Johnston and Alan Coffey for intellectual and critical input.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Massimo Gadina, PhD, is a Senior Lecturer and Oonah Lynch, PhD, Postdoctoral Fellow in the Department of Microbiology and Immunology at Queen’s University, Belfast. Their research focuses on signaling pathways that control lymphocyte activation, adhesion, and migration. Address correspondence to MG. E-mail m.g.gadina{at}qub.ac.uk; fax +44-(0)28-90325839

