The Environmental Genome Project
Phase I and Beyond
- National Institute of Environmental Health Sciences, National Institutes of Health, 111 T.W. Alexander Drive Research Triangle Park, NC 27709-2233
- Address correspondence to SHW. E-mail wilson5{at}niehs.nih.gov; fax 919-541-3592.
Abstract
Human illness is caused by many interrelated factors including aging, inherited genetic predispositions, and a variety of environmental exposures. There is increasing awareness of the role of genetics as a factor that can dramatically alter susceptibility to all disease, especially environmentally induced chronic disease, such as cancer, asthma, diabetes, cardiovascular disease, and neurodegenerative disorders. In some cases, a genetic factor influences disease susceptibility in a small fraction of the population because it occurs at a low frequency or involves a relatively low-incidence disease; however, in other cases, a genetic factor increases susceptibility in a large number of individuals and involves a disease that occurs at high incidence, creating a large public health burden.

Introduction
During the last half of the twentieth century, modern biology experienced dramatic discoveries and rapid technological developments that revolutionized biomedical research. This process began with the discovery of the DNA double helix, the breaking of the genetic code, and the development of recombinant DNA technology; eventually it led to innovations such as polymerase chain reaction (PCR) techniques, high-throughput DNA sequencing, and analysis of global gene and protein expression. From 1982 to 1998, DNA sequence information accumulated exponentially, as technology continued to improve and the cost of DNA sequencing decreased. Since 1998, DNA sequencing information has accumulated at an unprecedented rate, and we now realize that the human genome has a very large number of polymorphic sites, some of which are associated with susceptibility to disease. As the human genome project neared completion in the late 1990s, it became clear that a complete genome sequence would be an important tool for understanding human health and disease, although the sequence alone would be insufficient to answer many questions about the genetic basis of disease. In particular, an urgent need arose for a comprehensive database of genetic variation that would allow for the identification not only of highly penetrant (i.e., Mendelian) disease-associated genes, but also less penetrant loci that collectively contribute to disease. It would clearly be a large undertaking to create such a database of human DNA polymorphisms, but the biomedical imperative to do so was strong.
The mission of the National Institute of Environmental Health Sciences (NIEHS) is to reduce the harmful effects of environmental exposure—broadly defined to include physical, chemical, biological, behavioral, and social factors over time—on human health. Some of these factors are nutrition, hormonal status, infectious agents, industrial chemicals and by-products, radiation, socioeconomic status, lifestyle, behavioral patterns, and social stress. These environmental factors interact with genetic factors (1), so that the effects of environmental exposure can be complex and difficult to predict at the level of the individual (Figure 1⇓). The NIEHS has established several programs, including the Environmental Genome Project (EGP), to provide insight into the genetic component of environmentally induced human disease and to capitalize on recent technological advances in DNA sequencing and genomics-based science.
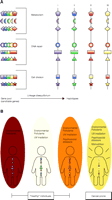
Human health as a function of the interrelationship between the genome and the environment. A. Eight hypothetical genes, involved in toxicant metabolism, DNA repair, or cell division, are represented as distinct geometries. Each of the eight genes can be found, in a given population, in five allelic variations (represented by five variations of color in the schematic). Due to linkage disequilibrium, however, the combinatorial variation of the eight genes in terms of alleles is limited; the combination of alleles that typifies an individual is referred to as a haplotype. In the schematized example, four haplotypes are seen to result in consideration of the eight genes selected for analysis (i.e., the eight candidate genes); for heuristic purposes, haplotype IV will be defined as a rare haplotype. B. A variety of environmental stressors can affect diseases, but the roles of individual candidate genes in these diseases may be difficult to assess. In the three environments indicated on the left, any of the four haplotypes may adequately respond to stress (although only one haplotype, selected at random, is shown in each environment), so that no disease phenotype is apparent; in this case, the term “wild-type” allele is purely arbitrary. In a fourth environment (yellow), a particular haplotype (here, the rare haplotype IV) may not be adequately robust (here, the yellow alleles are no longer visible) in the face of given stressors, in which case an association between “susceptible” alleles and disease may be made. The establishment of such associations is the ultimate goal of the Environmental Genome Project.
The EGP was formally initiated in 1997 as a comprehensive systematic approach to understanding how human genetic polymorphism influences individual susceptibility to environmentally induced disease (2, 3); thus, the EGP is an effort in functional genomics. It is a multicomponent effort composed primarily of investigator-initiated grants in five research areas: discovery of human genetic polymorphism; identification of functional genetic polymorphism; population-based epidemiological and clinical studies; technology development; and the ethical, legal, and social implications of genomics-based research (Box 1). There are currently more than 100 investigator-initiated projects among these research areas. This research will be used to develop and promote large-scale clinical and epidemiological studies that correlate single nucleotide polymorphisms (SNPs) and other DNA variations with disease risk. Ultimately, the EGP will help develop therapeutics, science-based environmental policy, and interventions that prevent illness from environmental exposure. Here, we describe and evaluate the accomplishments of the EGP to date.
Granting areas of EGF-sponsored research.
Sequencing projects include identification of DNA polymorphic sites, bioinformatics-based sequence analysis, validation of DNA sequence variants, comparative genomics, analysis of gene splicing, and studies of gene-regulatory regions. Population-based research projects include environmental, molecular, and pharmacogenetic epidemiolgy studies. This area also includes projects involving disease risk factor analysis, biomarker development, and SNP genotyping. Functional analysis projects focus on structure-function relationships, enzymology, subcellular localization, protein folding, tissue-specific gene expression, and transgenic and other animal model systems. Technology-focused projects are developing high throughput methods including DNA microarray technology and validation, protein mass spectroscopy, capillary electrophoresis, denaturing HPLC, and methods for whole cell protein analysis. Some technology projects are developing statistical methods to analyze gene-environment interactions and computational resources for analyzing raw data, databases, or macromolecular cellular components.
Discovery of Genetic Polymorphism
Many inherited disease traits with high penetrance have been identified using genetic linkage analysis. However, it has been much more difficult to identify genetic susceptibility loci, generally characterized by low penetrance, that are involved in common human diseases such as cancer, asthma, diabetes, cardiovascular disease, and Parkinson’s disease. These diseases are caused by several interacting genes as well as environmental factors, so that the relative risk for disease associated with a specific allele is expected to be very low. It is thought that individual susceptibility to these diseases is linked to the common DNA variations in the human genome. There are many types of DNA variations in the human genome, but the most common variations are SNPs. When two haploid genomes are compared, there is on average one SNP in 1,300 base pairs; there are four to five SNPs per gene coding region (i.e., cSNPs), and it is estimated that there are approximately 11 million SNPs in the human population (4); SNPs that occur at a frequency of 1% or greater are pursued for study by the EGP.
One approach to identifying SNPs is to analyze DNA sequences already deposited in databases (i.e., data mining). This approach does not provide SNP frequency information and is likely to underrepresent low-frequency SNPs. It is also possible to conduct large-scale resequencing of a human population sample to directly identify SNPs and estimate their population frequencies. This approach can be applied to the whole genome (given sufficient resources for such a large undertaking), to a targeted set of genes, or to specific genes in a targeted subpopulation. If the population sample and the set of targeted genes are appropriate, this approach yields valuable information that can be used to support clinical and epidemiological studies of gene–environment interactions. The EGP is taking just such a “candidate gene” approach using a carefully selected set of DNA samples (see below). In contrast, few other SNP discovery projects are using the candidate gene approach. To ensure the quality and usefulness of its SNP database, the EGP has implemented stringent quality control standards and validation procedures. In addition, the EGP has devoted considerable resources to enhance database management and maintenance mechanisms and to develop and apply analytical and computational tools that enhance the value of the GeneSNPs database.
Several organized efforts other than the EGP have been launched to create a comprehensive catalog of human SNPs. For example, the Five-Year Plan (1998–2003) of the Human Genome Project set a goal of identifying 100,000 human SNPs (5, 6). As part of this effort, the National Human Genome Research Institute and the National Center for Biotechnology Information established dbSNP, a public database for worldwide SNP data. dbSNP currently catalogs approximately 7.2 million SNPs and is built primarily through data mining obtained from sequence overlaps. Approximately 3.3 million SNPs are considered validated by multiple sequence analyses. The SNP Consortium Ltd. was initiated in 1999 as a nonprofit foundation whose initial goal was to create a public database of SNPs. The SNP Consortium includes academic centers and pharmaceutical companies, and the project has recently been expanded to determine allele frequencies and geno-types (7). The International HapMap Project was also launched in 2002 to study human haplotypes, which are sets of associated polymorphic sites (see below). These SNP databases will provide a resource for genetic association studies linking polymorphic variants (or combinations of variants) with human disease susceptibility.
The Candidate Gene Approach to SNP Discovery
The EGP’s candidate gene approach is designed as a long-term effort to systematically identify and characterize human genetic polymorphism in selected genes that are potentially involved in susceptibility to environmentally induced disease. An important aspect of the EGP’s SNP discovery effort was the selection of candidate genes. Because the candidate genes are fairly well characterized and in most cases have a significant probability of playing a role in disease susceptibility, the SNPs identified by the EGP have a good chance of being functionally important. The candidate genes targeted by the EGP are described further below; they are also listed on the GeneSNPs Web page (http://www.genome.utah.edu/genesnps) and on the EGP Web site (http://www.niehs.nih.gov/envgenom/home.htm).
Environmentally responsive genes tend to fall into eight categories: cell cycle, DNA repair, cell division, cell signaling, cell structure, gene expression, apoptosis, and metabolism. Cell cycle and cell division genes regulate the ability of a cell to proliferate, grow, and differentiate. Changes in the progression of a cell through the cell cycle can increase the ability of a cell to survive stress, for example, by allowing cellular damage to be repaired prior to cell division. Cell signaling and gene expression pathways have profound effects on all cellular functions, including cell proliferation and differentiation. Metabolic pathways are crucial determinants of the outcome of exposure. An inert innocuous compound can be metabolically converted into a reactive species that causes cellular damage; alternatively, some metabolic pathways destroy toxic compounds by changing a compound’s chemical structure. DNA repair genes influence the outcome of exposure to environmental agents that cause DNA damage. Individuals with higher or lower capacity for DNA repair have decreased or increased risk of certain types of environmentally induced disease, respectively. Heavily damaged cells often die by a process known as programmed cell death, or apoptosis. This process protects the organism by removing damaged or aberrant cells, and failure to execute the process is associated with adverse health effects, such as cancer.
The EGP has generated a list of 554 environmentally responsive genes as targets for resequencing in a large set of genetically diverse DNA samples representative of the U.S. population. The DNA samples are from ninety unrelated males and females including Americans of European, African, Mexican or Asian descent, and Native Americans. (More information about these and the full array of 450 DNA samples available from Coriell Cell Repositories can be obtained from http://locus.umdnj.edu/nigms/products/pdr.html.) EGP resequencing studies are designed to effectively detect polymorphic sites that occur at 1% or higher in the sampled population, by scanning the 180 chromosomes of the ninetysample subset of Coriell Cell Repositories’ polymorphism discovery resource. Because the frequency of a polymorphism can vary in different population subgroups, the EGP is sequencing DNA samples that represent the diversity of the U.S. population.
As with any large-scale and complex initiative, it was considered useful to break the timeline for the EGP into three broadly defined and partially overlapping phases (Table 1⇓). Phase I of the EGP was completed in the spring of 2003 and attained the following goals: identifying an appropriate set of DNA samples for polymorphism analysis; 2) resequencing 200 candidate genes; and 3) establishing a polymorphism database (see below). The following Phase-I EGP goals are relatively open-ended: 4) developing technology for identifying genetic variants in human genes; and 5) considering the ethical, legal, and social implications of EGP research. Although these goals were addressed prior to 2003, work on achieving these goals will continue during EGP Phases II and III. Phase II will also focus on resequencing genes regulating metabolism, signal transduction, and apoptosis. In addition to the ongoing EGP efforts, Phase II EGP goals include multidisciplinary functional studies of human genetic variants, population-based studies of exposure, and allelic associations; developing technology for Phase II studies includes toxicogenomics, additional mouse models, and refining the EGP SNP database. Progress on achieving these EGP Phase II goals has been quite rapid; two case studies are presented below as examples of EGP research activity related to these Phase-II EGP goals.
Phased Goals of the Environmental Genome Project
The GeneSNPs Database
A major accomplishment of the EGP is the compilation of the high-quality, publicly accessible GeneSNPs database, which is currently maintained at the University of Utah. The database is frequently accessed from among the general research community (≈35,000 visitors since September 2000), provides valuable SNP analysis and genotype visualization tools, and is actively curated. A summary of the types of genes targeted by the EGP resequencing study is shown in Table 2⇓. (Note that GeneSNPs includes data for genes other than EGP candidate genes.)
Summary of the GeneSNPs Database
Another primary goal of Phase I was the complete resequencing of 200 candidate genes; to date, approximately 300 genes have been resequenced. The majority of the Phase I genes were DNA repair or cell cycle genes; other sequenced genes (Phase I and Phase II) play roles in cell division, cell structure, signal transduction, metabolism, homeostasis, and gene expression. Approximately 30,000 new SNPs have been deposited in GeneSNPs, including more than 1,300 that appear in transcribed sequences and nearly 600 that potentially alter protein coding. The large majority of these SNPs (≈70%) had not appeared in the dbSNP database, which is consistent with the prediction that the dbSNP database (and other databases generated by data mining) are likely to underrepresent lower-frequency (<5%) SNPs. (The SNPs of the dbSNP database are distributed across the whole genome, but only a small fraction of the millions of SNPs are in the gene-coding sequences.) All SNPs in the GeneSNPs database have been validated by multiple sequence analyses and include genotype and frequency information. In contrast, less than half of the SNPs in the dbSNP database have been validated and only a small fraction include either genotype or frequency information. More than 300 genes (≈1% of all human genes) have now been sequenced in the EGP DNA resequencing effort, and close to a third of these genes were sequenced at >75% coverage (i.e., exons, introns, and regulatory regions). Partial data for an arbitrarily chosen subset of the EGP candidate genes are shown in Table 3⇓.
Examples of SNP Data from the GeneSNPs Database
One of the results emerging from analysis of SNPs in general is that they tend to associate with one another in a nonrandom manner known as linkage disequilibrium (8); associated SNPs (and other forms of associated genetic variation) are collectively referred to as a haplotype. On average, EGP candidate genes fall into twenty-five haplotypes, far short of the total number of possible SNP combinations. However, the number of haplotypes that typify any gene is highly variable: outliers have from 3 (FEN1) to 102 (CCND2) haplotypes. It is envisioned that haplotype and linkage disequilibrium data will simplify the task of large-scale genotyping, which is necessary for analyzing the genetic basis of disease by gene association studies. Thus, haplotype data are also being examined for most of the EGP candidate genes. The GeneSNPs Web site includes several tools for haplotype analysis (Figure 2⇓).
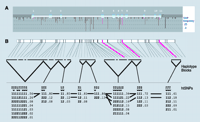
A GeneSNPs view of the methylenetetrahydrofolate reductase (MTHFR) gene. A. The MTHFR-encoding gene represents a typical EGP gene, in terms of size and nucleotide diversity, that has been scanned for polymorphism discovery. MTHFR has eleven exons (light blue indicates protein-coding and green indicates untranslated (UTR) sequences). For this gene, 15 kb was scanned for polymorphisms, which includes sequences 5′ to the first exon (~1.7kb) and 3′ of the last exon (~1.5 kb), by amplifying ninety U.S.-representative DNA samples using nineteen overlapping amplicons. Vertical descending lines represent SNPs identified in this sequence; the length of these vertical lines represents the frequency of the minor allele, and the color indicates whether the SNP location is in flanking (black), intronic (brown), synonymous (yellow), non-synonymous (red), or UTR (green) sequences. B. The structure of haplotype blocks along the MTHFR gene sequence is generated using the default algorithm from Gabriel et al. (31). For this gene, the algorithm partitions the SNPs with minor (>2%) allele frequencies into eight discrete blocks of linkage disequilibrium. Within each haplotype block are indicated the haplotypes, with the major and minor allele represented by a “1” or “2,” respectively. Population frequencies (>1%) are shown next to each haplotype block, and lines show the most common (>10%) recombination events between blocks. Marker numbers are shown across the top, with haplotype-tagging SNPs (htSNPs) highlighted with a triangular pointer.
EGP Case Studies
Functional Genomics of Paraoxonase(PON1) Polymorphisms
Polymorphisms in the paraoxonase gene (PON1) are associated with risk for vascular disease and with altered capacity to detoxify neurologically active substances including potential chemical warfare agents (9). PON1 is a high-density lipoprotein–associated plasma enzyme that metabolizes toxic organophosphates and some pharmaceutical agents (e.g., statins). During normal metabolism, the primary substrates of PON1 may be oxidized lipids, but PON1 plays an important role in the cytochrome P450 pathway that facilitates detoxification after exposure to organophosphate compounds.
EGP investigator Clement Furlong (University of Washington) conducted a detailed analysis of PON1 polymorphism and its impact on organophosphate sensitivity and cardiovascular disease. The human gene has two common SNPs in the coding region (resulting in L55M and Q192R substitutions), five promoter SNPs, one intronic polymorphic CA repeat, and four SNPs in the 3′ untranslated region. The L55M SNP does not alter enzyme activity or phenotype significantly, but the Q192R SNP has significant functional consequences. Homozygotes for the Q192R allele have more paraoxonase activity than heterozygotes or wild-type homozygotes. The increased activity is, however, substrate specific (10). The promoter region has three common haplotypes, which influence the amount of plasma activity. The promoter SNPs are in linkage disequilibrium with the L55M SNP. Recent analyses have identified a number of additional SNPs in both coding and noncoding regions that have not yet been well characterized.
The role of in organophosphate sensitivity was explored using a PON1−/− knockout mouse and knockout mice that had been injected with the human wild-type or Q129R PON1 (11). PON1- deficient mice die rapidly after exposure to chlorpyrifos oxon or diazoxon, whereas homozygous wild-type and heterozygous mice have high or moderate resistance, respectively. Surprisingly, PON1 genotype is not a determinant for paraoxon toxicity. Resistance to chlorpyrifos oxon or diazoxon was restored by injection of human, but resistance to paraoxon was not. Q192R PON1 provided much greater protection against chlorpyrifos oxon than the wild-type PON1, but the two isoforms protected equally well against diazoxon toxicity. PON1 activity also plays an important role in preventing oxidation of lipids and high-density lipoprotein (12). In a large set of carotid artery disease cases and controls, the presence of the Q192R allele did not correlate with disease susceptibility. However, the level of PON1 activity was significantly lower in individuals carrying at least one wild-type allele. This suggests that secondary factors (potentially promoter SNPs) may reduce PON1 activity in these individuals, which increases their susceptibility to carotid artery disease. This study clearly demonstrates that PON1 status has implications for susceptibility to environmentally associated diseases including organophosphate toxicity and cardiovascular disease. PON1 status also may influence susceptibility to Gulf War Syndrome. Future studies should also examine the roles of PON1, −2, and −3 in normal metabolism and in response to xenobiotic compounds.
Gene–Environment Interactions in Human Leukemia
EGP researcher Martyn Smith (University of California, Berkeley) has examined gene–environment interactions in blood-related cancers including leukemia, lymphoma, and myeloma. In the U.S., as many as 620,000 people are living with these cancers and 58,000 die from them per year. Blood-related cancers cause more deaths in children under fourteen years of age than any other disease. Of the four types of leukemia [i.e., acute myeloid (AML), chronic myeloid (CML), acute lymphocytic (ALL), and chronic lymphocytic leukemia (CLL)], AML is the most common form in adults and ALL is the most common in children.
Up to 20% of leukemia cases are thought to be induced by environmental factors, which include benzene, radiation, and chemotherapeutic agents. Genetic factors are also thought to play a significant role, especially in pathways controlling DNA repair and oxidative DNA damage. Identification of potential susceptibility factors could help narrow attention to those environmental exposures that increase leukemia rates. Possible genetic approaches include candidate gene analysis in individual patients, or in genomic DNA pools, or genome-wide scans of DNA pools.
Smith and collaborators identified one candidate gene that is likely involved in the etiology of leukemia, namely, the gene that encodes NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1). NQO1 plays an important role in preventing oxidative damage caused by exogenous and endogenous quinones. The common C609T polymorphism (occurring in 5–20% of the population) results in the P187S amino acid substitution, with a complete loss of enzyme activity in homozygotes. Case-control studies indicate a 1.5- to 2.5-fold increased odds-ratio for several types of leukemia in association with the 609T variant (13–15). This effect is relatively small, but adverse environmental exposure may interact with this genetic variant, leading to a significantly increased risk of disease.
Smith et al. have also investigated variation in genes involved in folate metabolism. Folate metabolism is critical to cell survival and stability, because it provides precursors for DNA synthesis, DNA repair, and methylation. Some of the critical enzymes in folate metabolism are methylenetetrahydrofolate reductase (MTHFR), serine hydroxymethyl transferase (SHMT), and thymidylate synthase (TS). There is a fork in the pathway, which shunts folate metabolites into DNA synthesis/repair (via TS) or methylation (via MTHFR). There are several functional SNPs that affect the enzymes of folate metabolism. These variants reduce leukemia risk 2- to 3-fold in heterozygotes and 3- to 10-fold in homozygotes (16, 17). Dietary folate also protects 2- to 3-fold against leukemia (18), and some evidence supports a role for genetic interactions between polymorphic variants of SHMT and TS (17).
Functional Genomics and Epidemiology in the EGP
The EGP’s catalog of human genetic polymorphisms will be the basis of valuable epidemiological research. However, the goal of validating the functional biological significance of human genetic variants is an extremely challenging long-term endeavor. In some cases, epidemiological evidence of functional biological significance will precede supporting molecular evidence. Epidemiological research that correlates polymorphisms affecting the DNA repair protein XRCC1 with cancer risk is a good example. In particular, female smokers with a specific XRCC1 SNP have an increased risk of pancreatic cancer (19), and combinations of two or more XRCC1 SNPs appear to contribute to breast cancer risk (20, 21). These and other results suggest a role for XRCC1 in cancer prevention. However, progress has been slow in understanding mechanisms that might explain these effects, even though some data suggest a role for XRCC1 in the DNA base excision repair pathway [for review, see (22, 23)]; thus, populationbased studies are ahead of the functional genomics of the SNPs in question. The fact that XRCC1 is essential during mouse embryogenesis (24) indicates that XRCC1 may play a conserved and essential biological role, and this role may relate to cancer prevention in mammalian tissue. Because the EGP is a cross-disciplinary research program, it will promote collaboration among epidemiologists, molecular geneticists, biochemists, and other scientists in an effort to define the biological role and mechanism of XRCC1.
Potential Concerns for the EGP Strategy
The DNA samples being resequenced through the EGP carry no sample-specific identifiers. Thus, EGP researchers are not able to search the database to obtain the precise frequency of specific alleles or haplotypes according to race, sex, or other subpopulation characteristics. Although such information could be valuable, concerns have been raised about artifacts that may occur in studies that use genetically distinct subpopulations to correlate allelic variation with disease (25). If multiple loci synergistically affect disease but only one locus is taken into account when comparing two subpopulations, the allelic consequences of the “background” genotypes may lead to erroneous conclusions about the locus under investigation. In such cases, better results will be obtained if samples from different populations are not combined during genetic analysis. Altshuler et al. (25) thus underscore the pitfall that it can be problematic to assume that a single genetic variant is associated with a single phenotype.
Some studies suggest that haplotype analysis can be more useful than the consideration of specific SNPs in establishing the genetic basis of disease (26). For example, Drysdale et al. showed that the efficacy of β2-adrenergic receptor (β2AR) agonists correlates better with β2AR haplotypes than with any single SNP found in a cohort of asthmatics (27). In addition, a recent paper reported that there are 4,304 haplotypes in a set of SNP data pertaining to 313 genes from eighty-two individuals including African Americans, Asians, European Americans, and Hispanics (28). The extent of allele sharing among individuals in the same population was only slightly greater than between individuals from different populations. Therefore, the concept that there is one predominant or “wild-type” form of a gene and various rare or “mutant” forms is simplistic. Instead, there are multiple haplotypes, each of which is observed in multiple populations, that account for a large fraction of human genomic variability (Figure 1⇑).
The Future of the EGP
The EGP has collected a significant amount of data on human genetic polymorphism in its first five years of activity. The rate of progress is expected to increase as the EGP closes in on the goal of analyzing SNPs in 554 environmentally responsive genes. In the next few years, the EGP will continue to investigate the functional significance of human genetic polymorphism, to improve SNP databases and technology that support research on genetic polymorphism, and to explore the ethical, legal, and social implications of research on human genetic polymorphism. EGP research in these areas is discussed briefly below.
GeneSNPs Enhancements
The GeneSNPs database will be enhanced with additional data on DNA structure, genotype information, and gene models and will provide improved visualization tools. In addition, we anticipate improving the capacity to organize disparate sequence data into coherent gene-based models. Several functional improvements to the database are planned, such as mapping amino acid changes onto predicted protein structures; more data on genotypes and haplotypes; comparative analyses of homologous human and mouse genes; population-specific allele frequencies; organization of gene lists into functional biological pathways and cellular networks; analysis of relevant gene and protein expression data; and improved database curation.
SNP Discovery
It is thought that most functional polymorphic sites lie in exons, but there are a few examples of functional variants that lie in introns (29). The long-term goal of the EGP is to resequence the complete genomic region for genes up to 25 kb. Genes larger than 25 kb will be “sample-sequenced” across the 5′ promoter region, 3′ untranslated region, protein-coding region, and all conserved non-coding sequences. In addition, limited sequencing will be carried out in large introns. These approaches will provide sufficient information for linkage disequilibrium analysis of these genes and will be useful for identifying haplotypes.
Functional Analyses
The EGP is involved in many projects that study the functional significance of human genetic polymorphism. One of the most ambitious of these projects is the Comparative Mouse Genomics Centers Consortium (CMGCC). This Consortium includes five Centers (i.e., Harvard University, University of Texas Health Science Center-San Antonio, University of Texas MD Anderson Cancer Center, University of Washington, and the University of Cincinnati) that are developing mouse models of human gene variants. Mutant mice will be created that carry the polymorphic sites found in human genes, and the functional effects of these variants will be assessed. Mutant mice will be subject to extensive phenotypic analysis including tests of susceptibility to environmental exposure. The first mouse models to be developed by the Consortium will be in DNA repair and cell cycle genes. Additional information about the Consortium can be found at the CMGCC Web site http://www.niehs.nih.gov/cmgcc/home.htm. In addition, the EGP is considering approaches to stimulate more widespread use of mouse models from the CMGCC and other sources in association studies of exposures, SNPs, and phenotypes.
Population-Based Studies
The NIEHS supports population-based studies to examine the distribution of SNPs in subpopulations. The EGP is also developing new statistical and computational models for analyzing gene–environment interactions and the means for analyzing data on macromolecular cellular components (e.g., DNA repair and recombination machinery). The EGP is working in close collaboration with the NIEHS National Center for Toxicogenomics (NCT). The NCT is using global expression technology, including microarrays and proteomics, to study the cellular response to environmental exposure. Microarrays, proteomics, and bioinformatics are used to identify molecular signatures that are associated with exposure and disease and to improve understanding of how genetic polymorphism influences susceptibility to environmentally induced disease.
Ethical, Legal, and Social Implications
Complex ethical, legal, and social issues are associated with research on human genetic polymorphism. NIEHS is collaborating with other NIH institutes, (e.g., National Human Genome Research Institute) to promote research in this area. Recently, NIEHS announced a new investigator-initiated program on “Environmental Justice: partnerships to address ethical challenges in environmental health.” This program will promote multidisciplinary community-based research. The involvement of community members and research scientists (30) is especially important in this area, so that the public can ultimately benefit from improved understanding of human genetic susceptibility. The ultimate goal of the EGP is to translate knowledge of human genetic polymorphism into mechanisms that prevent environmentally induced disease. Through concerted and long-term effort, and in collaboration with other groups who share this goal, it is hoped that the burden of human disease will decrease significantly.
Acknowledgments
The authors thank Miriam Sander for editorial assistance, Deborah Nickerson and Robert Weiss for critical review of the manuscript, and Jose Velazquez for discussions during early stages of its preparation. For their efforts in fostering the EGP, we thank Jose Velazquez, Joan Packenham, Elizabeth Maull, Leslie Reinlib, Kimberly Gray, Claudia Thompson, and Tommy Hardee. We are indebted to Deborah Nickerson, Mark Reider, Robert Weiss, Bruce Aronow, and Maynard Olsen for their contributions to the EGP databases. We thank Robert Weiss for assistance with Figure 2⇑.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Kenneth Olden, PhD, is Director of the National Institute of Environmental Health Sciences, National Institutes of Health.

Samuel H. Wilson, MD, is Deputy Director of the National Institute of Environmental Health Sciences, National Institutes of Health.

