MECHANISMS OF RADIATION INJURY TO THE CENTRAL NERVOUS SYSTEM: IMPLICATIONS FOR NEUROPROTECTION
Abstract
The central nervous system (CNS) is a major dose-limiting organ in clinical radiotherapy (XRT). The underlying mechanisms of radiation-induced injury in this organ remain unclear. For many years, research has focused on identifying the major target cells of damage, and depletion of target cells due to reproductive or clonogenic cell death was believed to be the primary cause of tissue damage and organ failure. There is now an increasing body of data indicating that the response of the CNS after XRT is a continuous and interacting process. This review addresses some of the recent advances in our understanding of the mechanisms of CNS radiation damage. Specifically, the focus is on apoptotic cell death, and cell death and injury mediated by secondary damage. These potentially reversible components of the injury response provide important targets for neuroprotective interventions.
Introduction
Radiation therapy (XRT) is a major cancer treatment modality. About 40% of all cancer patients receive XRT during their illness either for cure or for local control or symptomatic palliation (1). The effectiveness of XRT is, to a large extent, limited by the potential for normal tissue injury. Radiation injury of the central nervous system (CNS), consisting of the brain and spinal cord, has devastating clinical consequences. Because of its anatomic location, the CNS is not only dose-limiting in the treatment of CNS tumors, but also in the treatment of head and neck, thoracic, and upper abdominal malignancies. The underlying mechanisms of this injury remain unclear; however, there is an increasing body of data indicating that the response of the CNS after XRT is a continuous, dynamic, and interacting process (2). It is now recognized that clonogenic cell death is not the only mode of cell death in the CNS after XRT (3–8). Certain glial, neuronal, and endothelial cells in the CNS also undergo apoptosis soon after XRT. Furthermore, there is a component of secondary injury and cell death that is mediated by microenvironmental alterations such as hypoxia/ischemia and inflammation (9–13). This review addresses these potentially reversible components of the injury response and their implications on neuroprotective interventions.
Specific Cell Types in the Central Nervous System and Their Function
The major cell types in the CNS are neurons, glia, and vascular endothelial cells. Vascular endothelial cells in the CNS form the blood–brain barrier (BBB), essentially rendering the CNS impermeable to most proteins, hydrophilic molecules, and ions. Glial cells are generally divided into oligodendrocytes, astrocytes, and microglia. Oligodendrocytes are responsible for myelination of axons of neurons, a process that greatly increases the propagation of nerve impulses or action potential. Astrocytes participate in the transmission of the neuronal signals and in the formation and maintenance of the BBB. Microglia have phagocytic properties and are activated in inflammatory conditions of the CNS (14).
Clinical Radiation Therapy
Radiation injury to the CNS is manifest as three distinct clinical entities (15). During XRT, patients often experience fatigue and an increase in neurologic symptoms and signs, and death from brain herniation––especially after a large single dose, such as that delivered during stereotactic XRT––has been reported (16). The acute CNS radiation injury is generally considered to be secondary to edema and disruption of BBB (8, 17).
A self-limiting, delayed reaction known as Lhermitte’s syndrome is well recognized after XRT to the spinal cord. It occurs after a latent period of two to four months and is characterized by paresthesia in the back and extremities upon neck flexion. After cranial XRT, a corresponding syndrome characterized by somnolence has also been described (18). These syndromes typically last for a few months followed by complete clinical recovery. Transient demyelination is believed to be the underlying mechanism of these early delayed reactions.
Late effects, occurring after recovery from the early onset syndromes, are irreversible and most devastating, and thus are the most clinically important. Radiation injury to the spinal cord is associated with permanent motor and sensory deficits (15) and can be fatal if the damage occurs at the upper cervical level (19). In the brain, late effects include minor-to-severe neurocognitive deficits (20) and focal necrosis, the latter after higher doses (21). The systematic use of magnetic resonance imaging (MRI) leads to progressive and diffuse changes in white matter often after doses well below the threshold of necrosis (21, 22). The underlying histopathologic changes associated with these imaging findings and their relevance to the neurocognitive dysfunction observed after XRT are poorly understood.
Histopathologic Changes
Although few gross histopathologic changes are observed immediately after XRT, there are profound early changes at the cell and molecular level as discussed below. The histopathologic changes of late radiation-induced CNS injury include glial atrophy, demyelination and necrosis confined to white matter, and varying degrees of vascular changes in both white and gray matter (23). In the human CNS, these changes are typically described in the patients with the most severe injury (24), and human studies are also limited by confounding effects of many unknown host, treatment, and tumor factors. In clinical XRT, the radiation dose is typically given in a number of daily fractions over five to seven weeks. The influence of dose, fractionation treatment, time, and other treatment parameters on late functional and histopathological changes has been derived from studies in rodent models. For example, in XRT-associated paralysis of rats that occurs within seven months of XRT administration, necrosis and/or demyelination of the white matter occurs in the absence of apparent vascular abnormalities (23). Glial atrophy and vascular changes were observed much later and after lower doses. Late radiation-induced morphological changes in CNS microvessels include telangiectasias, dilatation, and vessel wall hyalinization and thickening with fibrinoid necrosis (25, 26). In the rodent spinal cord, these vascular changes are associated with no or very minor neurological deficits. In the brain, these vascular changes are associated with impairment of spatial learning and working memory, and were observed at doses below the threshold of necrosis (27). These histopathological studies provide important clues to the target cells of damage. For decades, a debate occurred regarding the role of the oligodendrocyte versus the vascular endothelial cell in the white matter lesions observed after XRT. With the availability of molecular tools and new insight in neurobiology, there has been a paradigm shift away from the target cell theory. Instead, the damage response is now increasingly viewed as a continuous, dynamic and interacting process.
Early Gene Induction
XRT produces a variety of DNA and other cellular lesions that elicit a stress response. Altered gene profiles are one characteristic feature of this response. In the rodent CNS, increased expression of pro-inflammatory and other genes has been demonstrated within hours following XRT (28–31). These include genes of transcription factors such as nuclear factor–kappa B (NF-κB), cytokines such as tumor necrosis factor–α (TNFα) and interleukin-1β (IL-1β), and basic fibroblast growth factor (bFGF). NF-κB is important in the regulation of cytokine expression, including expression of TNFα and IL1β, both of which have been implicated in the development of demyelination (32). The expression of TNFα is associated with edema observed after ischemic and hypoxic injury (33), and is also a key regulator of intercellular adhesion molecule-1 (ICAM- 1), which is associated with BBB disruption in a variety of injuries (34). Increased ICAM-1 expression has been observed in mouse brain and rat spinal cord within twenty-four hours after XRT (11, 35, 36).
In mouse brain after XRT, we observed an early dose- and time-dependent induction of ICAM-1 and heme oxygenase 1 (HO- 1). In animal models of impaired oxidative metabolism, induction of ICAM-1 paralleled the induction of HO-1 (which is a marker of oxidative stress) and subsequent neuronal death (37). Conversely, HO-1 modulates the expression of pro-inflammatory genes associated with endothelial cell activation (38). Modulating the expression of ICAM-1 and HO-1 may serve as a neuroprotective strategy against early radiation-induced BBB disruption and neuronal loss.
Similar to other CNS injury models associated with an increase in reactive oxygen species and a state of oxidative stress, the same was observed in the irradiated CNS. Amounts of malonaldehyde (MDA), an end product of lipid peroxidation, were elevated in mouse brain from six hours through seven days after XRT (17 Gy dose) to the whole cranium (unpublished data). MDA was elevated in the mouse hippocampus at one week after a dose of 10 Gy, and cells immunoreactive for MDA were observed in the dentate gyrus at twenty-four hours (39). Oxidative stress and increases in reactive oxygen species are likely to interact with radiation- induced altered gene profiles and to participate in responses to irradiation of the CNS.
Using cDNA microarrays, we have recently studied global gene expression in mouse brain during the first twenty-four hours after whole cranial XRT. XRT modulated the expression pattern of 192 genes (≥2.5-fold change), with sixty-three genes of known function and 129 as expressed sequence tags (ESTs). Gene expression profiles appeared to be dose- and time-dependent. Genes that were modulated at fifteen minutes after XRT were principally those involved in protein transport and biosynthesis, apoptosis, metabolism, or DNA repair. Those genes whose expression was altered at twenty-four hours post-treatment were generally involved in signal transduction, metabolism, cell cycle, and transcription (unpublished data). Real-time PCR of selected genes of interest confirmed similar expression changes after XRT, as had been identified by microarray analysis, which allows the identification of novel genes that may play a role in CNS radiation injury and offers the potential to elucidate novel molecular targets for neuroprotection.
Apoptosis
Certain cells undergo apoptosis within twenty-four hours after XRT, including oligodendrocytes (6, 7, 40), subependymal cells (which surround the ventricles of the brain and may include cells with properties of neural stem cells) (5, 41–43), certain neurons (4), and endothelial cells (3, 8). Neural precursor cells of the hippocampal dentate gyrus also are radiosensitive and undergo apoptosis within the first twenty-four hours after XRT (4). In rat spinal cord, we have shown that some of the apoptotic oligodendroglial cells have the phenotype of oligodendroglial precursors (unpublished data).
Radiation-Induced Apoptosis of Endothelial Cells
A 15% decrease in the number of endothelial cells in rat brain was documented twenty-four hours following a single X-ray dose of 25 Gy (44). A dose-dependent loss of endothelial cells at twentyfour hours was also observed in rat spinal cord after XRT (45). These findings are consistent with the observation of a peak of endothelial apoptosis at twelve hours in the rodent CNS post-XRT (3). Importantly, whereas oligodendrocyte apoptosis after XRT is p53-dependent (5), endothelial cell apoptosis is not p53-dependent. Radiation-induced endothelial cell apoptosis is mediated by the second messenger ceramide, which is generated from sphingomyelin following activation of sphingomyelinases (46). Thus, consistent with the notion that radiation-induced endothelial apoptosis in the CNS is mediated by the acid sphingomyelinase (ASMase) pathway, endothelial apoptosis after XRT was reduced in the CNS of ASMase knockout animals (3, 8).
Acute disruption of the BBB is well recognized after XRT of the CNS (45, 47–49). The dependence of radiation-induced endothelial apoptosis on the ASMase pathway provides a genetic approach to determine the role of endothelial cell apoptosis in acute BBB breakdown following XRT. Following a single dose of 50 Gy to the spinal cord, endothelial density was reduced by almost 50%, and disruption of the blood–spinal cord barrier (BSCB) was observed in ASMase wild-type mice, but not in ASMase-knockout animals (8). Both p53 wild-type and knockout mice exhibited a similar reduction in endothelial cell numbers and BSCB disruption after spinal cord XRT. These data provide compelling evidence that apoptosis of endothelial cells initiates acute BBB disruption in the CNS after XRT. One therapeutic implication is that targeting endothelial apoptosis may be a neuroprotective strategy against acute BBB disruption after XRT. As for early changes in gene expression, the relationship between early endothelial cell death and tissue injury that appears months later is not known.
Radiation-Induced Oligodendroglial Apoptosis
In the irradiated spinal cord, a reduction of oligodendrocytes twenty-four hours after XRT is observed (6, 40). At two to four weeks after XRT, there is a dramatic loss of oligodendroglial progenitor cells in rat spinal cord (50). After XRT of the rat cervical spinal cord with 15 or 23 Gy of 6 MeV photons, the clonogenic oligodendrocyte progenitor cell pool is reduced to less than 0.1% of its normal population, followed by a dose-dependent recovery, reaching a maximum of 40–80% of control values at three months post-XRT. Only after the paralytic dose of 23 Gy does a secondary decline of oligodendrocyte progenitors occur, between four and five months post-XRT (51).
Our recent data (unpublished) shows that loss of oligodendroglial progenitor cells was accompanied by glial proliferation. Proliferative cells have the phenotype of oligodendroglial progenitor cells. Early proliferation of oligodendroglial progenitor cells however was not impaired in the irradiated spinal cord of p53- knockout mice. Given the dependence of radiation-induced apoptosis of oligodendroglial cells on p53, this suggests that the early proliferation of oligodendroglial progenitors observed may not be linked directly to apoptosis of mature oligodendrocytes.
A number of early events, such as oligodendroglial apoptosis, reduced oligodendroglial density, and changes in oligodendroglial gene expression, were seen in the irradiated rodent spinal cord. These events were observed after a myelopathic dose of 22 Gy, and similar changes were observed after a much lower dose of 8 Gy. The relevance of early oligodendroglial apoptosis to late demyelination remains uncertain. These findings however suggest that these early events may not be directly associated with the late demyelination observed after XRT (40).
Inhibition of Neurogenesis
Neurons were once thought produced only during embryogenesis and soon after birth (perinatal). It is now accepted that neurogenesis persists in the adult CNS, and multipotent precursor or stem cells are present throughout adulthood (52). In the adult rodent brain, neurogenic stem cells are concentrated in the subventricular zone of the lateral ventricles and the subgranular zone of the hippocampal gyrus. There is evidence that neural stem cells may be a specific type of astrocytes residing within these regions (53, 54). There is a close association of neurogenesis with the hippocampal function of learning and memory, and newly generated neurons in the adult may participate in the formation of a hippocampaldependent memory (55, 56).
Recent studies suggest that inhibition of neurogenesis may contribute to the neurocognitive impairment after cranial XRT (9, 13, 57). In rat brain after a single XRT dose of 10 Gy, ablation of hippocampal neurogenesis occurs and neural precursors, instead, differentiate into glia cells. Transplants of non-irradiated neural precursor cells also fail to differentiate into neurons in the irradiated hippocampus. The inhibition of neurogenesis is accompanied by disruption of the microvascular angiogenesis associated with neurogenesis and an increase in the number and activation status of microglia (58), indicating that stem-cell differentiation is regulated by signals from the microenvironment in which these cells reside (54). Thus, in addition to clonogenic death of the neural progenitor cell population, the deficit in neurogenesis after XRT may reflect alterations in the microenvironment that regulates progenitor cell fate. These findings highlight the role of the microenvironment, in particular the presence of neuro-inflammation in mediating the damage response.
Rats with blocked neurogenesis following cranial XRT performed poorer than controls in hippocampus-dependent tasks (57). Although this finding suggests that the presence of newly generated neurons may be necessary for the intact hippocampus, it should be recognized that an enriched environment, exercise, and many other factors are associated with enhanced neurogenesis in rodents (59). The causal role of inhibition of hippocampal neurogenesis to the impairment of learning and memory function observed after XRT remains unproven.
Hypoxia, Barrier Disruption, and Necrosis of the White Matter
Many recent studies have focused on damage to the endothelial cells as a key target (15, 60). The importance of the vasculature was illustrated in boron-neutron capture therapy (BNCT) studies. Using capture agents to selectively irradiate the spinal cord microvasculature, histopathologic changes observed in rats that developed myelopathy after BNCT were virtually identical to those observed in animals treated with X-rays or neutrons only (61). These studies were confirmed by comparison of the surviving fraction of clonogenic oligodendrocyte progenitor cells after treatments with capture agents that did or did not cross the blood spinal cord barrier, or with treatment by thermal neutron beam only. The surviving fractions of clonogenic progenitors were significantly higher when the radiation dose was primarily delivered in the vascular endothelium, despite the fact that all treatments resulted in an equal incidence of white matter necrosis (51).
Consistent with this notion is the late disruption of BBB as a consistent finding that precedes gross white-matter damage in the CNS after XRT (62–64). We shall now focus on the disruption of the BBB and the associated micro-environmental changes that may lead to tissue damage.
Using the electron microscope to evaluate changes in endothelial cells in rat spinal cord, Stewart et al. noted a 30% reduction of microvessel endothelial density in the white matter, observed as early as three months after a single dose of 25 Gy (63). This observation was associated with focal disruption of the BSCB in white matter. It is unknown whether early endothelial apoptosis contributes to late microvessel density changes and BBB disruption. Failure of repair of early barrier disruption may also evolve into persistent barrier incompetence.
Hypoxia And VEGF
Hypoxia develops where oxygen supply from the vasculature is compromised because of deficient vascularization or local microcirculation disturbances, such as in ischemic or traumatic injury. Hypoxia causes a wide range of responses at both the systemic and cellular level, and has been proposed to regulate many physiological and pathological processes. Additionally, hypoxia is a crucial stimulus for vascular endothelial growth factor (VEGF)––also known as vascular permeability factor––which is known to mediate increased permeability in a wide range of tissues including the CNS (65). In the neonatal rat spinal cord, induced VEGF expression was observed within days and persisted for two weeks after a very high dose of XRT (55 Gy). This observation was followed by an increase in vascular density at four to five weeks after XRT (66). In the adult rat myelopathy model, where rats develop paralysis associated with necrosis of the white matter within twenty weeks after single doses of 20 to 25 Gy, a steep increase in the number of VEGF-expressing cells was observed beginning at sixteen weeks post-treatment. The increase in the number of VEGF-expressing cells also demonstrated a dose-dependent response above 17 Gy. The majority of these cells were astrocytes (67).
In a subsequent study (10), hypoxia in the irradiated rat spinal cord was assessed using two 2-nitroimidazole markers, [125I]- iodoazomycin arabinoside (IAZA) and 2-(2-nitro-1H-imidazol-1-l)- N-(2,2,3,3,3-pentafluoropropyl) acetamide (EF5), measured in the rat spinal cord using gamma-ray scintillation counting and immunohistochemistry, respectively. BSCB permeability was assessed using immunohistochemistry with an albumin-specific antibody and gamma-ray scintillation counting of [99mTc]-diethylenetriamine pentaacetic acid (DTPA). A dose-dependent increase in albumin staining and [99mTc]-DTPA activity beginning at sixteen weeks was observed, consistent with barrier breakdown. A similar dose-dependent increase in white matter astrocytes that showed immunoreactivity and in situ hybridization signals for VEGF was also observed. Irradiated rat spinal cord showed a dose- (17–22 Gy) and time-dependent (16–20 weeks after 22 Gy) increase in accumulated [125I]-IAZA compared to [125I]-IAZA accumulation non-irradiated controls. A similar pattern of dose- and timedependent EF5 immunoreactivity was also observed in white matter. Areas of EF5 expression and VEGF in situ hybridization signals co-localize with areas of albumin immunoreactivity. These results provided evidence for a dose-dependent temporal and spatial association of hypoxia, increased VEGF expression, and radiation- induced BSCB dysfunction.
To examine the functional consequences of altered VEGF expression, the response of VEGF-lacZ knock-in transgenic mice with increased or decreased functional VEGF expression to spinal cord XRT was assessed (12). Following XRT to the thoracolumbar spinal cord, transgenic mice with reduced VEGF showed protection and had a longer median time to development of weakness and paralysis compared to wild type mice and transgenic mice with increased VEGF. These results suggest a causal role for VEGF in the development of radiation myelopathy, and provide clear targets for intervention.
Hypoxia And Other HIF1-Target Genes
In the rat radiation myelopathy model, the number of astrocytes expressing hypoxia-inducible factor-1α (HIF1α), VEGF, and glucose-transporter-1 (Glut-1) increased with increasing doses of XRT above 17 Gy, and with increasing time after sixteen weeks following 20 Gy treatment. There was also spatial co-expression of HIF1α, VEGF and Glut-1 in regions of the spinal cord with evidence of hypoxia and BSCB disruption (12).
Reactive oxygen species are implicated in a number of degenerative and injurious processes in the CNS (68). They may play a role in CNS radiation injury (30). Various mechanisms may lead to the generation of free radicals under hypoxic conditions (69, 70). HO-1, a protein of oxidative stress, is induced by hypoxia through HIF1 (71). Upregulation of HO-1 in the rat spinal cord at 5 and 6 months after 26 Gy has been reported as evidence for oxidative stress (2).
Increased HIF1α may also upregulate the HIF1-target genes lactate dehydrogenase and other glycolytic enzymes such as phosphofructokinase, and aldolase-A. LDH induction is potentially harmful because it may lead to increased lactate production (72, 73). Whether this contributes to the altered microenvironment in the CNS under hypoxia remains uncertain.
Endothelial Tight Junction Proteins, Tight Junction Integrity, and BBB Disruption
The underlying mechanisms of VEGF-mediated increase in vascular permeability are unclear. There is evidence that VEGF-mediated vascular permeability changes do not require receptor binding (74). In radiation-induced late BBB disruption, VEGF upregulation was not associated with evidence for increased or even detectable VEGF receptor expression (12).
The integrity of the BBB is dependent on tight junctions, and possibly adherens junctions, between endothelial cells (75). VEGF increases the permeability of the brain endothelial cell monolayer and alters the expression and distribution of occludin and zonula occludens-1 (76). Incubation of umbilical vein endothelial cells with VEGF increased their permeability and decreased occludin expression (77). There is also evidence that hypoxia-induced increases in permeability of brain endothelial cells involve VEGFmediated changes in expression of zonula occludens-1 (78).
In a morphometric study using the electron microscope, no disruption or expansion of microvessel tight junction contacts was detected in the irradiated rat spinal cord (63). A different study demonstrated no apparent change in the distribution or amount of immunoreactivity of the tight junction proteins occludin and zonula occludens-1 in rat spinal cord after myelopathic radiation doses (11).
ICAM-1
The other class of proteins that are implicated in VEGF-mediated permeability increase is the adhesion molecules. Increased ICAM- 1 expression is associated with a diversity of CNS injury models where BBB disruption is present (79–81). Radiation-induced upregulation of adhesion molecules in the vasculature has been well documented for other organs (82). VEGF increases the expression of ICAM-1 in the CNS and in cultured brain microvascular endothelial cells (65, 83, 84).
ICAM-1 expression increases in white matter, but not in gray matter of rat spinal cord following a myelopathic dose of 22 Gy. The majority of glial cells expressing ICAM-1 appeared to be astrocytes. After 22 Gy, total ICAM-1 protein increased (at twentyfour hours post-irradiation), and was again elevated at seventeen to twenty weeks post-treatment. The time course, dose response, and spatial distribution of increased ICAM-1 expression paralleled the observations of BSCB disruption and albumin leakage (11). ICAM-1–mediated leukocyte binding, cytoskeletal rearrangements, and signaling to tight junctions may all contribute to barrier disruption (85, 86). Given the central role of ICAM-1 in the inflammatory status of endothelial cells, these findings are again consistent with the role of “inflammation” in propagating the CNS damage after XRT.
Targeting Damage Pathway For Neuroprotection
The data described suggest a model in which cell death and microenvironmental changes impacting cell fate and cell interactions contribute to tissue injury. We hypothesize that clonogenic cell death of endothelial cells following XRT initiates the initial breakdown of the BBB. This leads to vasogenic edema, ischemia, and hypoxia. Hypoxia induces HIF1-mediated increases in VEGF expression that, in turn, leads to a secondary damage cascade with further increase in vascular permeability, hypoxia, VEGF and ICAM-1 expression, and subsequent demyelination, and tissue necrosis. These finding have implications for neuroprotective strategies (Figure 1⇓).
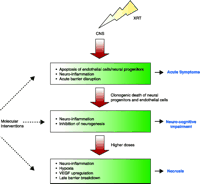
A model of molecular and cellular responses in the irradiated CNS. Neuroprotective interventions can be directed to potentially reversible targets of the injury response. Note that early injury might not be directly related to late neuro-cognitive impairment and necrosis.
Cell Replacement
In general, strategies for neuroprotection fall into two broad categories: 1) cell replacement, and 2) reduction in cell death. Cell replacement may be achieved through transplantation of stem cells or progenitors, or alternatively, through stimulation of proliferation or differentiation of endogenous stem cells or progenitors.
Ever since neural stem cells were isolated from the striatum of mouse brain (87), a large body of literature has grown describing their existence and fundamental properties. It is now accepted that neural stem cells persist in the adult brain and support ongoing neurogenesis in restricted regions within the CNS (53, 54). There is no unequivocal and unique marker for neural stem cells; these cells can only be identified definitively in vitro based on their multipotentiality and capacity for self-renewal; however, the possibility exists that neural stem cells may represent a special type of astrocyte within the subependyma.
For cell replacement therapy, neural stem cells from human fetuses are clearly unsatisfactory given their limited availability, issues of technical standardization and cell viability, not to mention ethical controversy. However, given the proper stimuli and environment, bone marrow stromal cells may differentiate into neural cells (88). They offer several advantages in terms of availability and lack of host immunity, and, importantly, they are readily obtained. Nonetheless, questions remain regarding whether differentiation of marrow stromal cells into neural cells represents the underlying mechanism for tissue repair. For instance, bone marrow stromal cells may instead create a milieu including angiogenesis that enhances regeneration of endogenous cells (89, 90).
The bone marrow has also been identified as a source of endothelial cell progenitors that are capable of homing to the CNS (91). Intravitreal injection of bone marrow–derived endothelial progenitors leads to normal retinal angiogenesis and can rescue pathologic vascular degeneration in the retina (92). The therapeutic potential of these endothelial progenitors in vascular degeneration is just beginning to emerge (93).
Transplantation of neural progenitors has been used to ameliorate radiation-induced myelopathy in rats (94). In this study, transplantation was performed by direct injection of immortalized neural stem cells into the irradiated spinal cord at day 90. Paralysis-free survival was improved in transplanted animals as compared to control animals. The fate of donor cells was not tracked in this study, and the biologic mechanisms for the efficacy observed remain speculative.
Stem cell self-renewal and progenitor differentiation is regulated by the microenvironment or niche in which these cells reside. Essential components of this niche may include signals for vasculogenesis and development (54). XRT may alter changes in the microenvironment such that the intrinsic factors or signals necessary for “proper” progenitor differentiation or self-renewal are no longer available. Thus, neural progenitors transplanted into irradiated rat hippocampus fail to develop into neurons and, instead, differentiate into glia (13). Furthermore, as more time is taken for transplanted stem cells to repopulate and spread to lesioned areas, there is increased detriment to these cells’ restorative potential (95). These and other complexities of stem cell therapies have been discussed elsewhere (96) and will not be detailed here. Much work is needed in the understanding of the biology of neural and vascular stem cells before transplantation or stem cell therapy can be considered as a neuroprotective strategy for radiation-induced CNS damage.
In addition to transplantation, cell replacement may be achieved through stimulation of proliferation of host stem cells or progenitors (97, 98). Stimulated proliferation of endogenous stem cells or progenitors within the irradiated environment is unlikely to be of benefit because clonogenic death is likely to be the most important mode of cell death after XRT treatment. The potential role of enhancing the proliferation of host stem cells or progenitors outside of the irradiated volume or bone marrow stromal cells remains to be investigated.
Reducing Apoptotic Cell Death
Protection against radiation injury may be achieved by reducing apoptotic endothelial cell death. Intravenous administration of bFGF immediately prior to XRT inhibits endothelial cell apoptosis in the murine CNS (3). Further study of key elements of the apoptotic pathway in this system may suggest growth factor –based (or other) approaches directed at reducing endothelial apoptosis. Inhibiting acid sphingomyelinase activity might provide a highly specific approach to reduce radiation-induced endothelial cell apoptosis. Indeed, acid sphingomyelinase inhibitors are currently being developed (99). It remains to be seen whether the challenge of modifying apoptosis in the important normal tissue compartments can be achieved without adversely impacting the ability to target and kill tumor cells.
Other growth factors that warrant further investigations include platelet-derived growth factor (PDGF) and insulin-like growth factor (IGF) (100). When PDGF was intrathecally administered concomitant with irradiation of the rat spinal cord, modest but significant protection was observed (101). Whether this was mediated through reduction of apoptotic cell death remains to be investigated.
Reducing Secondary Cell Death
The CNS, it has been argued, has a limited repertoire of responses to injury. The pathophysiology of CNS radiation injury shares many similar damage pathways with ischemic, inflammatory, and demyelinating insults. Our own data and emerging data from the literature are consistent with a model where alterations of the microenvironment (e.g., endothelial dysfunction, BBB disruption, microglial activation, and inhibition of neurogenesis) contribute to the final insult. Thus, another approach is to direct efforts at blocking or modifying damage-associated changes in the microenvironment. In this paradigm of injury, an important potential neuroprotector that has a pleiotropic function is erythropoietin (EPO).
Erythropoietin As Pleiotropic Neuroprotector
EPO has long been recognized for its central role in erythropoiesis. It is a glycoprotein produced by the kidney and is responsible for the proliferation, maturation, and differentiation of the precursors of the erythroid cells (102). Human recombinant EPO has been used for over a decade to treat anemia in uremic and cancer patients.
There is evidence that EPO may regulate neurogenesis. Mice lacking EPO receptors (EPOR) die at embryonic day 13.5 (E13.5). The fetal brains of EPOR−/− mice have reduced numbers of neural progenitor cells and extensive apoptosis (103). EPORs are expressed in the adult subventricular zone, and EPO infusion into the adult lateral ventricles results in decreased numbers of neural stem cells in the subventricular zone and an increase in newly generated cells migrating to the olfactory bulb (104). EPOR expression has been observed in neurons, astrocytes, and microglia (105).
The pleiotropic functions of EPO are now well recognized (106). Similar to its regulation in erythroid tissue, EPO within the CNS is inducible by hypoxia and is regulated by HIF1α (107, 108). Many cellular effects of EPO in the CNS have been described. They include inhibition of apoptosis, anti-inflammatory and antioxidative effects, prevention of glutamate-induced toxicity and stimulation of angiogenesis (109, 110).
There is now an emerging body of data on the neuroprotective effect of EPO against a wide variety of CNS insults (109). EPO infusion into the lateral ventricles of rodents prevents ischemiainduced learning disability and rescues hippocampal neurons from ischemic damage (111). EPOR is abundantly expressed at brain capillaries, and systemically administered EPO is able to cross the BBB to enter the brain (112). Indeed, systemic administration of EPO protects the rodent brain against ischemia-induced injury, concussive brain injury, immune damage in experimental autoimmune encephalomyelitis, and kainate-induced neurotoxicities (112). EPO also attenuates secondary inflammation in a rat spinal cord contusion model (113).
We have recently assessed hippocampal-dependent learning and memory function in mice after XRT using an eight-arm radial maze. At two and four months, mice given 17 Gy to the whole cranium showed a decrease in the number of correct entries and an increase in time to complete all eight entries. Animals irradiated to 17 Gy and given intraperitoneal EPO one hour after XRT showed no impairment compared to controls. EPO given alone without XRT did not influence performance at the radial maze, and locomotor activities were not impaired in mice at two and four months after XRT with or without EPO using open field test (114). Although the underlying mechanisms of these effects of EPO remain to be investigated, these results suggest that the neuroprotective role of systemically administered EPO may be applied to radiation injury.
EPO has been used extensively over the last ten to fifteen years for the treatment of anemia in cancer patients. Hence, any neuroprotective effects observed in preclinical models can be readily translated to the clinic. Similar to any biologic interventions, the potential adverse effect of EPO on tumor control needs to be investigated (115, 116).
Targeting Hypoxia and HIF1 Target Genes
Tissue oxygen conditions could be improved by increasing the amount of oxygen inspired. Despite some anecdotal reports of efficacy (117, 118), studies of hyperbaric oxygen in CNS radiation injury have not demonstrated a benefit (119). This may indicate a fundamental problem due to microvascular dysfunction; however, hyperbaric oxygen is normally used after tissue necrosis has developed to stimulate vasculogenesis. Alternatively, reoxygenation injury may paradoxically accelerate axonal injury and abrogate any potential benefits (120).
An alternative strategy is to direct intervention at hypoxiainduced genes or proteins, rather than at hypoxia or HIF1α. In addition to EPO, augmenting the expression of genes such as HO- 1 and Glut-1 may be of value. Enhancement of HO-1 responses might confer protection in hypoxia-induced pulmonary hypertension (121) and protect hypoxic cardiomyocytes against reoxygenation injury (122).
Transgenic mice with decreased VEGF activity were protected against radiation-induced myelopathy (12), suggesting that the inhibition of VEGF may confer protection. Barrier disruption may also provide a window of access for small molecules administered systemically to inhibit VEGF. Many available inhibitors of VEGF block receptor activation (123, 124). However, permeability increases that are associated with VEGF might not require its binding to the VEGFR. VEGF induces phosphorylation of tight junction proteins occludin and zonula occluden 1 (125). Other strategies thus may include modulating the effects of VEGF on tight junction proteins. VEGF-mediated increases in vascular permeability are associated with endothelial nitric oxide synthase (eNOS) activity and release of nitric oxide (NO) (83). Inhibition of NOS activity might therefore be used to reduce vascular dysfunction.
Targeting Neuroinflammation
XRT is associated with induction of inflammatory markers including cytokine expression (28, 31). An increase in cyclooxygenase-2 (COX-2) expression and COX-2-mediated prostanoid production was observed in the irradiated mouse brain (17, 31). COX-2 is one of two isoforms of the obligate enzyme in prostanoid synthesis and a principal target of non-steroidal anti-inflammatory drugs (NSAIDs). Inhibition of COX-2 attenuates prostanoid induction and cerebral edema in mice after XRT (17).
XRT-induced inhibition of neurogenesis accompanies inflammation in the microenvironment and activation of the microglia (13). Indomethacin (an NSAID agent) given daily after XRT decreases microglial activation and restores neurogenesis within the subgranular zone of the dentate gyrus (9). The administration of indomethacin or other NSAIDs normalizes vascular permeability in intracranial gliomas (126). Reducing the inflammatory status of endothelial cells––as measured by expression of chemokines and ICAM-1––may represent the underlying mechanism of neuroprotection. Indomethacin may also exert its neuroprotective effect through mechanisms related to NO neurotoxicity rather than through COX-2 inhibition (127).
Increased trafficking of neutrophils into the CNS has been described for a number of injury models. In rat brain, increases in leukocyte–endothelial cell interactions were seen twentyfour hours and also three weeks after XRT in association with decreased blood flow (128). In a rat brain window model, the increase in adherent leukocytes was associated with increase in BBB permeability (49). Enhanced leukocyte trafficking has largely been ascribed to the adhesion molecule ICAM-1. The expression of ICAM-1 is induced in mouse brain and rat spinal cord after XRT (11, 35, 36, 129); it thus follows that increased leukocyte trafficking into the CNS may exacerbate the inflammation and BBB disruption induced by XRT injury.
Using a monoclonal antibody or another antagonist to block the extracellular interactions of ICAM-1 at the vessel lumen, one might be able to interrupt pathways activated by the binding of leukocytes, fibrinogen, or other interactions with as yet unknown binding partners. Injection with an ICAM-1–specific monoclonal antibody reduces leukocyte adhesion and BBB disruption in the rat cranial window model (49). Following thoracic XRT, ICAM-1 and inflammation contribute to pulmonary fibrosis and injury (130), and expression of VCAM-I and ICAM-I in the irradiated mouse lung was decreased by manganese superoxide dismutase– plasmid/liposome gene therapy (131). These findings identify targeting adhesion molecules as a potential molecular intervention to reduce neuroinflammation.
Conclusion
Recent studies have contributed significantly to our understanding of the molecular and cellular events that underlie the development of CNS radiation injury. Radiation-induced clonogenic cell death of “target cells” may not represent the sole mechanism of tissue injury. Apoptotic cell death and cell death arising from secondary injury may contribute to the development of long-term CNS injury. Specifically, cytokine cascades and stress responses associated with the damage deserve further investigations. These potentially reversible components of cell death present many targets for interventions using pharmacologic and biologic agents.
Acknowledgments
Studies in the laboratory of CSW reported here were supported by the National Cancer Institute of Canada. We thank Ms. Heather Bird and Su Horn for the technical support in manuscript preparation.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Albert J. Van der Kogel, MD is Professor of Radiobiology and Head of the Laboratory of Experimental Radiotherapy at the Department of Radiation Oncology, University Medical Center in Nijmegen, The Netherlands. For thirty years his research has focused on the effects of radiation on the central nervous system. During the last decade his group has mainly worked on clinical and experimental research of the tumor microenvironment, with emphasis on imaging and modifying tumor hypoxia.

C. Shun Wong, MD, is the Chief of Radiation Oncology at Sunnybrook and Women’s College Health Sciences Centre, and a Professor in the Departments of Radiation Oncology and Medical Biophysics, University of Toronto. He has a long-term interest in the responses of the central nervous system to ionizing radiation. Address correspondence to CSW. E-mail shun.wong{at}sw.ca; fax 416-217-1338.

