Purine Receptors: GPCR Structure and Agonist Design
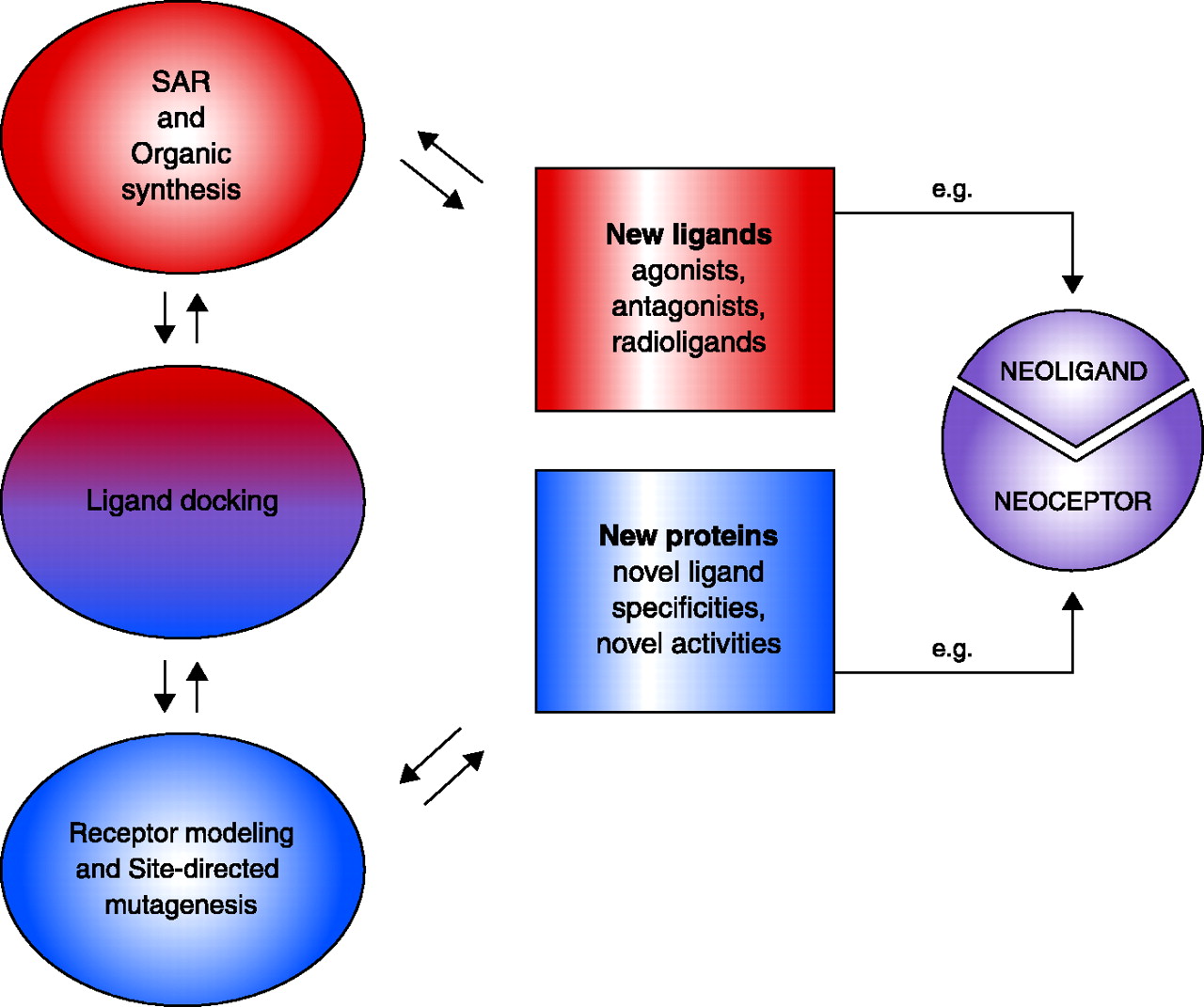
Integration of drug design and receptor modeling. Experimental data [e.g. structure-function relationships (SAR)] reveal interactions between multiple sets of ligands and receptors (i.e., both wild-type and mutant proteins); rational refinement of receptor (by mutagenesis) and ligand design (by organic synthesis) can then be suggested by ligand docking. The two stages—ligand design (red) and receptor design (blue)—are combined in an iterative manner to culminate in new ligands (e.g., agonists, antagonists, and new pharmacological probes) that will selectively bind, fail to bind, or modulate wild-type or mutant receptor proteins. A special integration of the two stages can culminate in “neoligands” that selectively bind to engineered “neoceptors.” It should be noted that GPCR modeling currently depends on the high-resolution structure of bovine rhodopsin (resting state) as template (4).

