Histamine in Cardiac Sympathetic Ganglia: A Novel Neurotransmitter?
Historically, cardiac ganglia have been considered collections of cholinergic neurons that distributed most densely near the sinoatrial and atrioventricular nodes (1–3). However, it is now generally accepted that cardiac ganglia contain a heterogeneous population of neurons capable of synthesizing, and responding to, several different neurotransmitters and neuropeptides including adrenergic and purinergic agonists and antagonists (2). Recently, Li and colleagues (4) as well as others (2, 5) have proposed that histamine may in fact meet the definition of a neurotransmitter in the heart. However, whether histamine be classified as a true neurotransmitter in cardiac sympathetic ganglia is less clear.
Histamine produces a wide array of effects in the heart (6–8). In fact, the effects of histamine on cardiac function have been appreciated since the work of Dale and Laidlaw (9) in 1910 who showed that synthetic histamine, β-imidazolylethylamine, modified cardiac rhythm in the mammalian heart. Subsequently, the effects of histamine and G protein–coupled histamine receptor subtypes on cardiovascular function have been well characterized [reviewed in (8, 10)]. Three of the four histamine receptor subtypes are present in the heart: H1 and H2 are located postjunctionally, whereas the H3 receptor is a prejunctional synaptic receptor (11, 12). A fourth histamine receptor, the H4 receptor, is widely expressed in hematopoietic cells (13–15) but to date has not been reported in the heart.
Histamine elicits multiple effects in the heart, including an increase in sinus rate, as demonstrated in transmembrane action potential recordings in sinoatrial (SA) nodal cells, increased ventricular automaticity (Box 1) through an H2-mediated enhancement of inward Ca2+ current (ICa), and subsequent acceleration in phase 4 spontaneous depolarization (Box 2) (16–18). The subcellular mechanisms by which histamine increases the slope of spontaneous diastolic depolarization and thereby augments the firing rate of SA nodal cells includes stimulation of adenylyl cyclase and increased adenosine 3′,5′-monophosphate (cAMP) formation and the activation of protein kinase A (PKA) which produces Ca2+ channel phosphorylation and augmented Ca2+ influx. Also, histamine profoundly decreases atrioventricular (AV) conduction velocity––an effect that is mimicked by H1 agonists, antagonized by H1 receptor blockade (19), and has been observed in multiple species, including humans (8, 20). Although activation of H1 receptors stimulates phosphoinositide turnover and increases intracellular cGMP in the myocardium, the precise mechanism of H1-mediated reductions in AV conduction has not been fully delineated (8). In spite of decreasing AV conduction velocity, histamine may actually increase AV nodal automaticity via H2 receptors through a mechanism analogous to that described for increases in sinus automaticity (8) and that has been demonstrated in dogs following suppression of the sinus rhythm (21) and in isolated, blood-perfused, AV nodal preparations from dog (22).
Definitions of Cardiovascular Terms
-
Automaticity and the Sinus Node: Automaticity refers to the propensity for a cell to elicit an electrical impulse on its own. A single specialized location in the atria, the sinoatrial node, has a higher automaticity (i.e., a faster pacemaker) than the rest of the heart, and therefore is usually the one to start the electrical impulse resulting in a heartbeat. The sinus fibers connect directly with the atrial fibers, so that any action potential that begins in the SA node spreads immediately into the atria.
-
AV Conduction Velocity: The speed with which the electrical impulse transmits from the atrial tissue and through the atrioventricular (AV) node to the Purkinje fibers and ventricular tissue of the heart.
-
Purkinje Fibers: Conducting fibers within the ventricle responsible for the rapid transmission of the electrical impulse throughout the ventricle.
-
Inotropy: The term applied to changes in the force of heart muscle contraction (e.g., a positive inotropic agent elicits an increase in the force of ventricular contraction whereas a negative inotrope produces a reduction in the force of contraction).
Phases of the Ventricular Action Potential
-
Phase 0: Rapid depolarization of the cardiac cell first mediated by INa and later by activation of ICa resulting in rapid contraction of the ventricle.
-
Phase 1: Activation of transient outward K+ current, Ito, that is present in subepicardial ventricular cells but to only a small extent in the remaining ventricular myocardial cells.
-
Phase 2: Plateau phase of the action potential predominately mediated by L-type Ca2+ channels resulting in the sustained contraction of the heart.
-
Phase 3: Repolarization of the cardiac cell, predominately mediated by K+ channels, resulting in ventricular relaxation.
-
Phase 4: Maintenance of the resting membrane potential to allow for filling of the heart prior to the next contraction. Modified from (41).
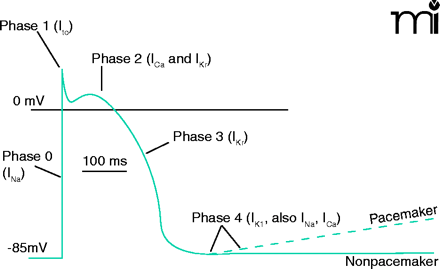
Importantly, histamine increases the force of ventricular contraction. However, the receptor subtype, or subtypes, that mediate this effect are unclear and in fact, the coupling between histamine receptor subtypes and the transduction mechanisms responsible for positive inotropic effects varies considerably among species and also for different regions of the heart [reviewed in (8)]. In the human, however, H2 receptors mediate the positive inotropic action of histamine in both atria and ventricles because increases in contractile activity are invariably blocked by cimetidine, but not pyrilamine (an H1 receptor antagonist) (23).
Finally, the release of cardiac histamine from both mast cell and non–mast cell sources during anaphylaxis or myocardial ischemia, or in response to drug treatment (e.g., anthracycline antibiotics, morphine, and d-tubocurarine) is known to provoke arrhythmogenic effects on the sinus node, atrial fibers, AV node, Purkinje fibers, and ventricular cells owing to changes in normal automaticity and conduction [reviewed in (24)]. In fact, in experimental models of acute myocardial ischemia, concentrations of histamine in the coronary sinus increase concomitant with the development of early ischemia-induced ventricular arrhythmias and in proportion to their severity (25); thus, histamine receptors in the heart may represent novel therapeutic targets.
Regardless of the well-characterized effects of histamine in the heart, whether histamine is truly a neurotransmitter in cardiac sympathetic nerve fibers necessitates the question, what defines a neurotransmitter? Schwartz (26) defines a neurotransmitter by four distinct criteria: 1) it is synthesized in the neuron, 2) the molecule is present in the presynaptic terminal and is released in amounts sufficient to exert a defined action on the postsynaptic neuron or effector organ, 3) when administered exogenously it mimics the action of the endogenously released transmitter, and 4) a specific mechanism exists for removing the molecule from the synaptic cleft. Several labs have taken even greater strides to more precisely define a neurotransmitter (4, 27) and add that, in addition to being released in response to neuronal depolarization, the transmitter release must be Ca2+ dependent. Further, blocking the release of the substance (i.e., histamine) should prevent presynaptic impulses from altering the activity of the postsynaptic cells and postsynaptic cells must bear the appropriate receptors for the substance. Therefore, these seven pre-defined and comprehensive criteria can be used to address the question regarding histamine function as a potential neurotransmitter in cardiac sympathetic ganglia.
Do cardiac sympathetic neurons synthesize histamine and is histamine present in nerve terminals?
Singh and colleagues (2) demonstrated that histamine-synthesizing enzymes are present in the neurons of adult human cardiac ganglia. They showed that 40% of neurons were immunoreactive for histidine decarboxylase, the key enzyme in histamine synthesis and that the enzyme was located in the neuronal soma and in dendritic and axonal processes. Evidence also suggests that histamine is stored in cardiac ganglia; Singh et al. (2) demonstrated, in human adult hearts, that almost 50% of neurons in the cardiac ganglia were immunopositive for histamine. Although Singh et al. (2) did not demonstrate the colocalization of histamine and NE to the same neurons in adult human cardiac ganglia, Li et al. (4) recently suggested that histamine and NE do coexist in 51% of cardiac sympathetic varicosities within guinea pig ventricles, although the evidence for the coexistence of the two substances in the neuronal soma is less convincing. Thus, cardiac sympathetic nerves appear to express the appropriate enzymes to synthesize and to store histamine intraneuronally prior to release.
Are histamine receptors present in the heart or on pre- or post-synaptic neurons?
Three of the four of the known histamine receptors appear to be expressed in the heart or on presynaptic sympathetic ganglia (11). In human tissue, evidence from both Northern and Western blot studies have indicated a variable distribution of H1 and H2 receptors and suggest that the relative distribution of the receptors may mediate the functional responses to histamine (23). H3 receptors also appear to be expressed on sympathetic ganglia in the heart although receptor mRNA or protein expression has not been conclusively demonstrated in cardiac synaptosomes or tissue. Despite the lack of physical evidence of H3 receptors in cardiac sympathetic ganglia, Imamura and colleagues (12) have functionally characterized these receptors on presynaptic cardiac nerve endings. They suggested that H3 receptors were present on sympathetic nerves in the human heart using H3-selective agents and demonstrated that the receptor is involved in the inhibition of K+-stimulated norepinephrine (NE) release and electrically-induced inotropic responses. Therefore, three histamine receptor subtypes have been physically or functionally characterized on either presynaptic neurons or in postsynaptic cells of the heart itself (11) and can respond to neuronal histamine release.
Is histamine release from presynaptic terminals Ca2+-dependent?
Li and colleagues (4) demonstrated that depolarization of cardiac synaptosomes (50 mM K+) stimulates modest (0.3–0.6 pM) but highly variable (± 0.4 pM) increases in endogenous histamine release, an effect that is not affected by Compound 48/80, a mast cell histamine-releasing agent, suggesting that cardiac histamine likely originates from a sympathetic neuronal source. Also, they demonstrated that the N-type Ca2+ blocker ω-conotoxin but not lacidipine, an inhibitor of L-type Ca2+ channels, attenuated histamine release, suggesting that endogenous histamine release may be dependent on N-type Ca2+ channel activation. Although the modest and variable increases in histamine release following depolarization were statistically significant, as were the reductions in histamine release in the presence of ω-conotoxin, the relevance of these small changes in synaptic histamine concentrations under physiological conditions is less clear.
Are sufficient quantities of histamine released to exert electrophysiological effects on the heart or in pre- or post-synaptic neurons?
Gross and colleagues (28) and Imamura et al. (29) have demonstrated that sympathetic stimulation of isolated guinea pig hearts produces a 1.5- to 3-fold increase in histamine overflow into the coronary perfusate. However, the concentrations of histamine under normal physiological conditions may be insufficient to activate sensitive H3 receptors because thioperamide, an H3 blocker, does not modify the resultant tachycardia or NE overflow (i.e., excess output of NE from neurons) following sympathetic activation, whereas thioperamide did increase NE overflow under conditions of ischemia and reperfusion. Thus, under physiological conditions, presynaptic H3 receptors in the heart may be quiescent whereas endogenous histamine does likely play an important role in the regulation of NE release under pathological conditions including myocardial ischemia.
Does exogenous histamine mimic the action of the endogenously released histamine?
Exogenous histamine (10 μL, 100 μM), when applied adjacent to spontaneously active canine right atrial neurons in situ increases neuronal activity and when administered into the local arterial blood supply of these neurons in vivo (100 μL, 100 μM) increases neuronal activity (from 8 ± 1 to 34 ± 4 impulses/min) and produces elevations in heart rate (from 119 ± 3 to 134 ± 4 beats/min) and right and left ventricular intramyocardial systolic pressures (from 106 ± 6 to 120 ± 7 mm Hg). Thus, exogenous histamine might activate a population of cardiac neurons relevant to cardiovascular function (30). These results are consistent with the effects of endogenous histamine in the heart as elucidated by selective pharmacological blockade or stimulation of specific histamine receptor subtypes (as detailed earlier). Also, both endogenous and exogenous histamine exert similar effects on NE release from cardiac sympathetic ganglia in experimental models (31–33) and in cardiac synaptosomes isolated from human atria (12). Endogenous NE release during ischemic stress has been linked to cardiac arrhythmias, and the modulation of NE release by presynaptic H3 receptor activation has been well documented (12, 33) and shown to reduce arrhythmia severity in vivo independent of changes in coronary flow (10, 34, 35). Luo et al. (36) first demonstrated the ability of H3 receptors to modulate sympathetic neurotransmission using isolated guinea pig hearts whereby (R)-α-methylhistamine, a selective H3 agonist, inhibited the positive inotropic effects of electrical field stimulation. More recently, Li and colleagues (4) demonstrated that K+-evoked NE release was attenuated in cardiac synaptosomes preloaded with L-histidine, an effect reversed by thioperamide, further suggesting H3 involvement. Also, using H3-selective agents, Silver et al. (31) and Seyedi et al. (37) collectively demonstrated that H3-mediated reductions in NE exocytosis from cardiac sympathetic nerves results sequentially from H3-receptor Gi/Go coupling, inhibition of adenylyl cyclase activity, and decreased cAMP formation, leading to a reduction in PKA activity and decreased Ca2+ influx through N-type Ca2+ channels. Thus, evidence suggests that under certain conditions both exogenous and endogenous histamine modulates cardiovascular function directly through H1 and H2 receptors and also modulates sympathetic NE release via stimulation of presynaptic H3 receptors.
Will blockade of histamine release prevent activity in presynaptic cells or effector organs?
Li and colleagues (4) suggest that histamine induced activation of presynaptic H3 receptors by (R)-α-methylhistamine results in the attenuation of K+-evoked histamine release from cardiac synaptosomes (from 3.75 ± 0.57 pM to 2.73 ± 0.68 pM) and suggest that the H3 receptor is effectively an autoinhibitory receptor in cardiac sympathetic neurons; how the blockade of histamine release from cardiac ganglia subsequently affects cardiovascular function was not investigated. However, others demonstrated that (R)-α-methylhistamine can modulate NE release in isolated guinea pig atria and attenuates the inotropic and chronotropic responses to adrenergic nerve stimulation (33). Although histamine release was not measured, inhibition of the adrenergic-induced chronotropic response was attenuated by the H3-inhibitor thioperamide and exacerbated in the presence of H1 and H2 receptor blockers, suggesting that the blockade of histamine release from sympathetic ganglia does antagonize the typical chronotropic effects elicited by histamine at H2 receptors (33).
Is histamine actively eliminated from the synaptic cleft?
Two possible pathways could effectively limit histamine concentrations in the synaptic cleft: 1) histamine transport across the membrane of pre- or post-synaptic cells or 2) histamine degradation within the synapse. Histamine may be removed from the synapse by the high-capacity vesicular monoamine transporter 2 (VMAT2), and because the H3 receptor may potentially function as a presynaptic autoinhibitory receptor that modulates the release of not only NE but also of histamine itself (4), presynaptic H3 receptors may effectively reduce histamine concentrations in the synaptic cleft. Whether the H3 receptor truly functions as a clearance receptor, however, to limit histamine concentrations in the synapse has not been conclusively demonstrated. Moreover, although the major histamine-metabolizing enzyme, histamine N-methyltransferase, is present in the neuron (4), the presence of this enzyme or of histaminase has not been observed in the synaptic cleft of sympathetic cardiac ganglia. Thus, although histamine may be transported from the synaptic cleft by VMAT2, it is not clear whether H3 receptors effectively limit histamine concentrations at the synaptic cleft or whether enzymes exist in the synapse of cardiac sympathetic ganglia to selectively degrade histamine.
When the available evidence is assessed using a comprehensive definition of a neurotransmitter based on seven distinct criteria, histamine appears to fall short of classification as a neurotransmitter in cardiac sympathetic ganglia. Clearly, histamine meets some of these criteria (Figure 1⇓). It appears to be synthesized and stored in the neurons and its receptors are present on both presynaptic cells and in the heart itself (although conclusive physical evidence of presynaptic H3 receptors in cardiac sympathetic ganglia has not been shown). Limited evidence also suggests that histamine release may be dependent upon N-type Ca2+ channel activation but whether sympathetic ganglia in the heart actively release histamine to concentrations sufficient to exert effects under physiological conditions is less clear. On the other hand, endogenous histamine does appear to play an important role in the modulation of cardiac function during pathological states (e.g., ischemia), and pharmacological studies have suggested that the effects of endogenous histamine on cardiovascular function and the regulation of NE release are similar to that of exogenous histamine. Selective modulation of histamine receptors clearly affects cardiovascular function in vivo and NE exocytosis in synaptosomes, thus it may be hypothesized that the blockade of histamine release would prevent this activity in postsynaptic cells; however, the actual blockade of postsynaptic activity in the presence of attenuated histamine release has not been conclusively demonstrated. Moreover, with the exception of the possible functional absorption of histamine in the synapse by presynaptic H3 receptors or by VMAT2 transport, there is no known mechanism to selectively eliminate or degrade histamine from the synaptic cleft of cardiac sympathetic ganglia.
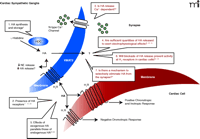
Does histamine meet seven distinct criteria in cardiac sympathetic ganglia that define a neurotransmitter? Histamine (HA) appears to be synthesized by histidine decarboxylase (HDC) and also stored within cardiac ganglia (1). G protein–coupled histamine receptors are present on both presynaptic cells and in the heart itself (2); however, only limited evidence suggests that histamine release may be dependent upon N-type Ca2+ channel activation (3). Whether sympathetic ganglia in the heart actively release histamine to concentrations sufficient to exert effects under physiological conditions is less clear (4), although endogenous histamine may play an important role in the modulation of cardiac function during pathological states. Pharmacological studies have suggested that the effects of endogenous histamine on cardiovascular function and the regulation of NE release are similar to that of exogenous histamine (5); however, the blockade of endogenous histamine release to abolish activity at presynaptic H3 receptors or at postsynaptic H1 and H2 receptors has not been conclusively demonstrated (6). Finally, with the exception of the possible functional absorption of histamine in the synapse by presynaptic H3 receptors or VMAT2-mediated transport, there is no known mechanism to eliminate selectively or to degrade histamine from the synaptic cleft of cardiac sympathetic ganglia (7).
Regardless of the classification of histamine as a neurotransmitter or simply as an important signaling molecule in the heart, histamine receptors do offer novel therapeutic targets in cardiovascular disease. The H1 and H2 receptor subtypes are expressed differentially across types of cardiac tissues in a species-dependent manner (23), accounting for the pharmacological and functional differences seen with histaminergic agonists and classical anti-allergy H1 receptor and anti-ulcerogenic H2 receptor antihistamines in these tissues. In humans, the predominant subtype in both normal atrial and ventricular tissues is the H2 receptor (23), which when activated appears to produce a positive inotropic response. Histamine induces arrhythmogenic activity in diseased human heart tissue (16, 24), and in conditions of endotoxemia, expression of both H1 and H2 receptors are increased, leading to the augmented effect of histamine in cardiac tissue (38). Thus, it is conceivable that selective H2 receptor antagonists may be of benefit in such disease states. Interestingly, the H2 receptor antagonist famotidine has also been shown to lessen the severity of chronic heart failure, perhaps suggesting a new mode of treatment for this disease (39).
The H3 receptor has received considerable interest as a potential target for cardiovascular diseases in recent years because of its presynaptic role in the modulation of the release of cardiac neurotransmitters including norepinephrine. Myocardial ischemia is associated with overstimulation of the sympathetic system, enhanced norepinephrine release, resultant dysrhythmias and metabolic demand, and aggravation of the initial ischemic event that can lead to additional heart damage and failure (10, 40). In fact, a greater incidence and longer duration of ventricular fibrillation, which was correlated with norepinephrine overflow, was seen upon reperfusion in hearts from H3 knockout mice subjected to ischemia (34). Thus, activation of H3 receptors by H3 agonists with drug-like properties may offer a novel and attractive means for the treatment of myocardial ischemic arrhythmias.
In conclusion, the present literature does not definitively support the classification of histamine as a neurotransmitter in cardiac sympathetic ganglia. Promising therapeutic opportunities exist, however, for subtype-selective histamine receptor agents in several cardiac diseases ranging from the prevention of heart failure and ischemia-induced ventricular arrhythmias to the blockade of hemodynamic consequences that result from histamine overload during endotoxemia. doi:10.1124/mi.6.1.3
Acknowledgments
The authors gratefully acknowledge Dr. Roberto Levi for his important suggestions and critical review of this manuscript.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Ryan M. Fryer, PhD, (right) is an Associate Research Investigator in Global Pharmaceutical Research and Development at Abbott Laboratories in the Department of Integrative Pharmacology. Please address correspondence to RMF. E-mail ryan.fryer{at}abbott.com; fax (847) 938-5286.
Glenn A. Reinhart, PhD, (middle) is a Senior Group Leader in Global Pharmaceutical Research and Development at Abbott Laboratories in the Department of Integrative Pharmacology.
Timothy A. Esbenshade, (left) PhD, is a Senior Group Leader in Global Pharmaceutical Research and Development at Abbott Laboratories in the Department of Neuroscience.

