Resveratrol in Cardioprotection: A Therapeutic Promise of Alternative Medicine
Abstract
Resveratrol, a polyphenol phytoalexin, possesses diverse biochemical and physiological actions, including estrogenic, antiplatelet, and anti-inflammatory properties. Several recent studies determined the cardioprotective abilities of resveratrol. Both in experiments (acute) and in chronic models, resveratrol attenuates myocardial ischemic reperfusion injury, atherosclerosis, and reduces ventricular arrhythmias. It appears that resveratrol-mediated cardioprotection is achieved through the preconditioning effect (the best yet devised method of cardioprotection), rather than direct protection. Thus, resveratrol likely fulfills the definition of a pharmacological preconditioning compound and gives hope to the therapeutic promise of alternative medicine.

Evolution Of Discovery To Cardioprotection
Almost 4500 years ago, Ayurveda, the ancient medicinal book of Hindus described “darakchasava” (fermented juice of red grapes) as cardiotonic (1). Consequently, The Bible described grape juice or red wine as a “gift of god,” which was presumably used to purify body and soul. In 1940, resveratrol was first identified as the medicinal component of grapes, and it was extracted from the dried roots of Polygonum cuspidatum (popularly known as Ko-jo-kon in Japan) and used to treat hyperlipidemic diseases (2). In the modern era, resveratrol has been rediscovered as an antiproliferative agent for cancer (3, 4). The cardioprotective ability of resveratrol stemmed from epidemiological studies indicating that mild-to-moderate alcohol consumption is associated with reduced incidence of morbidity and mortality from coronary heart disease (5). The first experimental evidence of cardioprotective ability of resveratrol was apparent from studies showing that resveratrol could directly protect isolated hearts from ischemia reperfusion injury (6). Subsequently, resveratrol was found to protect most of the vital organs including kidney, heart, and brain from ischemic reperfusion injury (7–9). The protective mechanism of resveratrol includes its role as intracellular antioxidant, anti-inflammatory agent, its ability to induce nitric oxide synthase (NOS) expression, and its ability to induce angiogenesis (10). A substantial body of evidence strongly supports the notion that resveratrol mediated cardioprotection is achieved by preconditioning; the best yet devised method of myocardial protection (11, 12).
Sources Of Resveratrol
The richest source of resveratrol is the roots of Polygonum cuspidatum (Ko-jo-kon), mainly cultivated in China and Japan. The skins of grapes contain about 50–100 mg/resveratrol, and believed to be responsible for the cardioprotective properties of red wine, which contains about 0.2–7 mg/l of wine. In addition to grapes, a large variety of fruits including mulberry, bilberry, lingonberry, sparkle-berry, deerberry, partridgeberry, cranberry, blueberry, and jackfruit, peanut, and a wide variety of flowers and leaves including gnetum, white hellebore, corn lily, butterfly orchid tree, eucalyptus, spruce, poaceae, scots pine, and rheum also contain resveratrol. Resveratrol is synthesized in response to environmental stressors that include water deprivation, UV irradiation, and, especially, fungal infection. Thus, the production of resveratrol in plants can be considered part of the defense mechanism.
A Natural Antioxidant
Resveratrol is capable of scavenging some intracellular reactive oxygen species (ROS). Although it possesses antioxidant properties, reseveratrol does not function as a strong antioxidant in vitro (10). Resveratrol can scavenge hydroxyl radicals with a reaction rate constant of 9.45 x 108 M−1sec−1, which is slower than the potent scavenging demonstrated by ascorbic acid (13). Depending on the biological system, resveratrol can also scavenge the superoxide anion (O2−) (13–15).
Although resveratrol behaves as a poor in vitro scavenger of ROS, it functions as a potent antioxidant in vivo. The in vivo antioxidant property of resveratrol probably arises from its ability to increase nitric oxide (NO) synthesis, which in turn functions as an in vivo antioxidant, scavenging superoxide radicals. In the ischemic reperfused heart, brain, or kidney, resveratrol induces NO synthesis and lowers oxidative stress (11, 16). Having an unpaired electron, NO behaves as a potent antioxidant in vivo, rapidly reacting near the diffusion-limited rate (6.7 x 109 M−1s1) with O2− that is presumably formed in the ischemic reperfused myocardium. The affinity of NO for O2− is far greater than the affinity of superoxide dismutase (SOD) for O2−. In fact, NO may compete with SOD for O2−, thereby removing O2− and sparing SOD for other scavenging duties.
The effects of resveratrol have been tested in specific cells lines and differentiated cell types. For example, resveratrol inhibits 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced free radical formation in cultured HL-60 cells (17). In DU 145 cells––a prostate cancer cell line––administration of resveratrol inhibited proliferation that had been accompanied by a reduction in NO production and inhibition of inducible nitric oxide synthase (iNOS) (18). Resveratrol also inhibited the formation of O2− and H2O2 produced by macrophages stimulated by lipopolysaccharide (LPS) or TPA (15). In a related study, resveratrol inhibited reactive oxygen intermediates and lipid peroxidation induced by tumor necrosis factor (TNF) in a wide variety of cells (19). Resveratrol also scavenges peroxyl and hydroxyl radicals in the postischemic reperfused myocardium, thereby lowering malonaldehyde formation, a presumptive marker of lipid peroxidation (6, 20).
Resveratrol can maintain the concentrations of intracellular antioxidants found in biological systems. For example, resveratrol maintained glutathione (GSH) amounts in oxidation-stressed peripheral blood mononuclear cells isolated from healthy humans (21). In another study, resveratrol increased GSH amounts in human lymphocytes that were activated by hydrogen peroxide (22). Similarly, resveratrol restored glutathione reductase in cells subjected to TPA-mediated oxidative stress (23). In human lymphocytes, resveratrol increased the amounts of several antioxidant enzymes, including glutathione peroxidase, glutathione-S-transferase and glutathione reductase (24).
A Phytoestrogen
Resveratrol has been recognized as a phytoestrogen based on its structural similarities to diethylstilbesterol (DES). Resveratrol can bind to the estrogen receptor (ER), thereby activating transcription of estrogen-responsive reporter genes transfected into cells (25–31). Resveratrol functions as a superagonist when combined with estradiol (E2) to induce the expression of estrogen-regulated genes (25); however, several other studies showed conflicting results. Using the same cell line, Ashby et al. showed that resveratrol possessed antiestrogen activity because it suppressed progesterone receptor expression induced by E2 (32). Another recent study showed that both isomers of resveratrol possessed superestrogenic activity only at moderate concentration (>10 μM), whereas at lower concentrations (<1 μM), antiestrogenic effects prevailed (33).
Most of the in vivo studies have failed to confirm the estrogenic potential of resveratrol. At physiologic concentrations, resveratrol does not appear to induce any changes in uterine weight uterine epithelial cell height, or serum cholesterol (34). Only at very high concentrations does resveratrol interfere with the serum cholesterol–lowering activity of E2. Elsewhere, resveratrol given orally or subcutaneously did not affect uterus weight at any concentration (0.03 to 120 mg/kg/day) (32). In a related study, resveratrol reduced uterine weight and decreased the expression of ER-α mRNA and protein and progesterone receptor (PR) mRNA (35).
In contrast, resveratrol possesses estrogenic properties in stroke-prone spontaneously hypertensive rats (36). When ovariectomized rats were fed resveratrol at the concentration of 5 mg/kg/day, treatment attenuated an increase in systolic blood pressure. In concert, resveratrol enhanced endothelin-dependent vascular relaxation in response to acetylcholine and, in a manner similar to estradiol, prevented ovariectomy-induced decreases in femoral bone strength. Resveratrol also acts as an estrogen receptor (ER) agonist in breast cancer cells stably transfected with ERα (37). Although more data accumulate on the estrogenic behavior of resveratrol, the controversy continues to persist.
Health Benefits Of Resveratrol
As mentioned earlier, several recent studies determined the cardioprotective abilities of resveratrol. Both in acute experiments and chronic models of cardiac injury, resveratrol reduced myocardial ischemic reperfusion injury (38, 39). In addition to hearts, resveratrol also protects kidney and brain cells from ischemia-reperfusion injury. Similar to its effect on the heart, resveratrol’s stimulation of NO production during ischemia-reperfusion is believed crucial for protection of the kidney from ischemic reperfusion injury (40). The maintenance of constitutive NO release is a critical factor in the recovery of function after an ischemic injury. Release of constitutive NO is significantly reduced after ischemia-reperfusion, thus the maintenance of NO release by any means (e.g., induction of NO production with l-arginine) can restore myocardial function following ischemia (41) (Figure 1⇓).
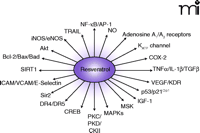
Molecular Targets of Resveratrol. A number of signaling pathways related to inflammation and gene expression are modulated by resveratrol.
Although resveratrol protects brain, kidney, and heart cells, it preferentially kills cancer cells. For example, intraperitoneal administration of resveratrol increased apoptosis and reduced tumor growth (42). In oral squamous carcinoma cells, resveratrol inhibited growth; both alone and in combination with quercetin, an antioxidant found in the skin of apples and red onions (43), and inhibited the growth of highly metastatic B16-BL6 melanoma cells (44). In a rat colon carcinogenesis model, resveratrol induced the expression of the proapoptotic protein Bax in aberrant cryptic foci of the colon (45). In fact, resveratrol affects three major stages of carcinogenesis (i.e., initiation, promotion, and progression) and inhibits the formation of preneoplastic lesions in a mouse mammary organ culture model (4).
Resveratrol inhibits the growth of cytotoxin-associated gene–carrying (CagA+) strains of Helicobacter pylori in vitro (46) as well as fifteen other strains of H. pylori (47), suggesting that its presumed bacteriostatic activity might be part of its overall salutary effects. Other beneficial effects of resveratrol include its ability to increase (13-fold) the activity of the anti-aging gene SIRT1 [the mammalian homolog of the S. cerevisiae gene silent mating type information regulation 2 (Sir2)] (48). Thus, resveratrol-mediated activation of life-extending genes in human cells may open a new horizon of resveratrol research (Figure 2⇓).
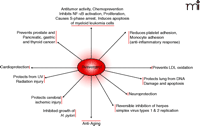
Health benefits of Resveratrol. Reveratrol has been implicated as a therapeutic in a number of major diseases
Cardioprotection With Resveratrol
Vasorelaxation
The role of NO in the cardioprotective abilities of grape products first became apparent when certain wines, grape juices, grape skins, or their components were found to relax precontracted smooth muscle of intact rat aortic rings but had no effect on aortas in which the endothelium had been removed (49). The extracts of wine or grape increased guanosine 3′,5′–monophosphate (cGMP) amounts in intact vascular tissue, and both relaxation and the increase in cGMP were reversed by NG-monomethyl-l-arginine or by NG-nitro-l-arginine––competitive inhibitors of the synthesis of the endothelium-derived relaxing factor, NO––suggesting that vasorelaxation induced by grape products is mediated by the NO-cGMP pathway. A direct role for NO in vasorelaxation was identified when increased NOS activity was found in cultured pulmonary artery endothelial cells treated with resveratrol (28), suggesting that resveratrol could afford cardioprotection by affecting the expression of NOS. Astringinin, an analog of resveratrol, also exhibits NO-mediated beneficial effects during ischemia and reperfusion damage in rat heart (50). Pretreating rats with astringinin significantly dampened the incidence ventricular tachycardia (VT) and ventricular fibrillation (VF) and decreased the concentrations of lactate dehyodrogenase (LDH) found in the carotid artery. Consistent with these results, resveratrol protected perfused working rat hearts through increased iNOS expression (11). The cardioprotective ability of resveratrol was abolished with an iNOS inhibitor, aminoguanidine. Resveratrol also failed to provide cardioprotection in iNOS knockout mice (51). In a recent study, however, resveratrol reduced myocardial ischemia reperfusion injury through both iNOS-dependent and iNOS-independent mechanisms by increasing the expression of iNOS, endothelial NOS (eNOS), and neuronal NOS (nNOS) (52).
Anti-inflammatory Effects
The cardioprotective ability of resveratrol also stems from its anti-inflammatory functions in the ischemic heart. Treatment with the polyphenol resveratrol significantly improved postischemic ventricular function and reduced myocardial infarct size compared to non-treated control group (53). The amount of proadhesive molecules, including soluble intracellular adhesion molecule-1 (sICAM-1), endothelial leukocyte adhesion molecule-1 (sE-Selectin), and vascular cell adhesion molecule-1 (sVCAM-1) were each significantly decreased during reperfusion in the resveratrol-treated group. Nitro-l-arginine methyl ester (l–NAME), an NO blocker, completely abolished such beneficial effects of resveratrol. The results support an anti-inflammatory action of resveratrol through a NO-dependent mechanism.
The anti-inflammatory role of resveratrol is also evident from a research where resveratrol effectively suppressed the aberrant expression of tissue factor (TF) and cytokines in vascular cells (54). Resveratrol, in a dose-dependent manner, inhibited the expression of TF in endothelial cells stimulated with a variety of agonists, including interleukin-1β (IL-1β), tumor necrosis factor–α (TNF-α), and lipopolysaccharide (LPS). Nuclear run-on analyses in endothelial cells showed that resveratrol inhibited the transcription of the TF gene. In another recent study, resveratrol attenuated the agonist-induced increase of TF mRNA in endothelial and mononuclear cells, resulting from the inhibition of IκBα degradation, thus decreasing the DNA-binding occupancy by the transcription factor c-Rel/p65 (55). Elsewhere, resveratrol inhibited platelet aggregation induced by collagen, thrombin, or ADP (56). Additionally, trans-resveratrol interfered with the release of inflammatory mediators by activated polymorphonuclear cells (PMN) and decreased the adhesion-dependent thrombogenic functions of PMN (57). ADP-induced platelet aggregation in hypercholesterolemic rabbits can be blocked by treatment with resveratrol, suggesting a role for resveratrol in interrupting the generation of eicosanoids involved in pathological processes. Indeed, Pinto et al. showed that resveratrol could inhibit the dioxygenase activity of lipooxygenase and that oxidized resveratrol was as efficient as the reduced form in inhibiting lipoxygenase (58). Similarly, trans-resveratrol inhibited, in a dose-dependent manner, the arachidonate-dependent synthesis of the inflammatory agents Thromboxane B2 (TxB2), hydroxyheptadecatrienoate (HHT), and 12-hydroxyeicosatetraenoate (12-HETE) (59).
ROS Scavenging
Resveratrol protects the heart against ROS-mediated menadione (i.e., Vitamin K3) toxicity by inducing NAD(P)H:quinone oxidoreductase, also known as DT-diaphorase, a detoxifying enzyme for quinone-containing substances (60). The cardioprotective effect of resveratrol was also attributed to its ability to upregulate catalase activity in the myocardium. Resveratrol functions as in vivo antioxidant and can scavenge peroxyl radicals in the heart (61, 62). A commercial preparation of resveratrol made from P. cuspidatum root extract (Protykin®) also scavenges peroxyl radicals and protects the heart from ischemia reperfusion injury (63).
Anti-Apoptosis
Resveratrol appears to induce an anti-apoptotic signal for the protection of the heart. In porcine coronary arteries, short term treatment with resveratrol significantly inhibited mitogen-activated protein kinase (MAPK) activities, and immunoblot analyses revealed consistent reduction in the phosphorylation of extracellular signal-regulated kinases 1/2 (ERK1/2), Jun N-terminal kinase (JNK-1), and p38 MAPK (64). The same study found that resveratrol attenuated basal and endothelin-1 (ET-1)-mediated protein tyrosine phosphorylation. Anti-apoptotic function of resveratrol is further supported by several other studies, which have demonstrated a reduction of apoptotic cardiomyocytes in the ischemic reperfused heart that had been pre-treated with resveratrol (51, 63) (Figure 3⇓).
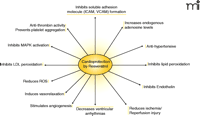
Cardioprotection with resveratrol. The major contribution to health from resveratrol appears to be directed toward limiting progression of heart disease and atherosclerosis. See text for details.
Pharmacological Preconditioning With Resveratrol
Preconditioning is the most powerful technique known to promote cardioprotection (64–69). The most generalized method of classical preconditioning is mediated by cyclic episodes of several short durations of reversible ischemia, each followed by another short duration of reperfusion. In most laboratories including our own, preconditioning is achieved by four cycles of ischemia (five minutes each) followed by ten minutes of reperfusion (66, 70). Such preconditioning makes the heart resistant to subsequent lethal ischemic injury.
The mechanisms underlying cardiac preconditioning have been studied extensively, and several regulatory pathways have been identified in different systems. Three important factors: the adenosine A1 receptor, multiple kinases [including protein kinase C (PKC), MAPKs, and tyrosine kinases], and the mitochondrial ATP-sensitive potassium (KATP) channel are known to play a crucial role in preconditioning-mediated cardioprotection. For example, cardioprotection achieved by preconditioning can be abolished by A1 receptor antagonists (71, 72), whereas A1 receptor agonists can limit myocardial infarct size (73). Although there is general agreement regarding the beneficial role of adenosine on ischemic tissue, Berne’s hypothesis of adenosine-mediated regulation of coronary blood flow (i.e., the adenosine hypothesis) remains controversial (74). This hypothesis is inconsistent with several findings that inhibition of KATP channel blocks the effects of preconditioning and that a KATP channel opener can simulate ischemic preconditioning (73, 75). To reconcile the adenosine hypothesis, an argument has been made that adenosine could trigger a secondary mechanism such as activation of Gi protein, which, in turn, could open the KATP channel.
Another intriguing hypothesis has stemmed from the concept of stimulating an endogenous protective mechanism by myocardial adaptation to ischemic stress. Preconditioning induces the expression of endogenous antioxidant enzymes such as superoxide dismutase (SOD) and glutathione peroxidase (GSHPx-1) (76, 77) and the heat shock proteins (HSP) HSP27, HSP32, and HSP70 (78, 79). Additionally, preconditioning potentiates a signal transduction cascade by inhibiting death signals and activating survival signals. Thus, several proapoptotic and antiapoptotic genes and transcription factors, including JNK-1, c-Jun, NF-κB and AP-1, are likely to play a crucial role in preconditioning (80–82), as does NO (83, 84).
Recent studies from our laboratory determined that resveratrol may function as a pharmacological preconditioning agent as it fulfills several criteria for preconditioning, including activation of adenosine A1 and A3 receptors, PKC, MAPKs, and the KATP channel. Adenosine accumulates in tissues under metabolic stress. In myocardial cells, the nucleoside interacts with various receptor subtypes (i.e., A1, A3, and perhaps A2A and A2B) that are coupled, via G proteins, to multiple effectors, including enzymes, channels, transporters and cytoskeletal components. Studies using adenosine receptor agonists and antagonists, as well as animals overexpressing the A1 receptor indicate that adenosine exerts anti-ischemic action. Adenosine released during preconditioning, by short periods of ischemia followed by reperfusion, induces cardioprotection to subsequently sustained ischemia. This protective action is mediated by A1 and A3 receptor subtypes and involves the activation and translocation of PKC to sarcolemmal and to mitochondrial membranes, leading to PKC-dependent KATP channels. Other effectors possibly contributing to cardioprotection by adenosine or preconditioning that seem particularly involved in the delayed (i.e., second) window of cardioprotection (Box 1) include MAPKs, heat shock proteins, and iNOS and eNOS. Because of its anti-ischemic effects, adenosine has been tested as a protective agent in clinical interventions such as percutaneous transluminal coronary angioplasty (PTCA), coronary artery bypass grafting (CABG), and tissue preservation, and was found in most cases to enhance the post-ischemic recovery of function. The mechanisms underlying the role of adenosine and of mitochondrial function in preconditioning are not completely clear, and uncertainties remain concerning the role played by newly identified potential effectors such as free radicals, the sarcoplasmic reticulum, etc. In addition, more studies are needed to clarify the signaling mechanisms by which A3 receptor activation or overexpression may promote apoptosis and cellular injury.
The Two Phases of Cardioprotection
Acute or classical preconditioning can produce marked reduction of myocardial infarct size and cardiomyocytes apoptosis in all species tested so far; however, its clinical potential is limited because of its short duration of approxiamtely 2–3 hours. A delayed protective effect reappears after 12–24 hours, following early preconditioning and this cardioprotective effects last up to 72 hours.
Several investigators have proposed a unifying hypothesis that activation of PKC represents a link between cell surface receptor activation and a putative end-effector sarcolemmal or mitochondrial KATP channels (85, 86). Possible involvement of protein tyrosine kinases in preconditioning was proposed for the first time by Maulik et al. (87). Now, it is increasingly clear that protein tyrosine kinases play a crucial role in mediating preconditioning in some animal species (88). Protein tyrosine kinases may act in parallel to (89, 90), downstream of (91, 92) or upstream of (93) PKC in eliciting preconditioning; however, the identity of these protein tyrosine kinases remains unclear. Among a large number of tyrosine kinases, Src family tyrosine kinases have received much attention (94). Src has been implicated in the mechanism of cell survival and death, which is regulated by complex signal transduction processes (95). Rapid activation of Src family tyrosine kinases after ischemia has also been documented in the isolated guinea pig heart (96). Members of the Src family can be activated by many effectors, including stimulation of G protein–coupled receptors (97), increased intracellular Ca2+ (98), oxidative stress (99), and enhanced nitric oxide synthesis (84), all of which can be elicited by preconditioning challenges.
The intracellular signaling mechanisms that mediate preconditioning require the activation of one or more MAPKs. Among these, the stress-activated protein kinases (SAPKs, also termed JNKs, see above) and p38 are known to be regulated by extracellular stresses, including environmental stress, oxidative stress, heat shock, and UV radiation (100). JNKs and p38 appear to be involved in distinct cellular functions, because they possess different cellular targets and are located on different signaling pathways. Thus, JNKs activate c-Jun whereas p38 stimulates MAPK-activated protein kinase 2 (MAPKAP kinase 2) (101). A recent study demonstrated that preconditioning triggered a tyrosine kinase-regulated signaling pathway that leads to the translocation and activation of p38 and MAPKAP kinase 2 (87).
Activation of KATP channels appears to be an adaptive mechanism that protects the myocardium against ischemic reperfusion injury (102). The activation of this ion channel is at least partially responsible for the increase in outward K+ currents, shortening of action potential duration (APD), and increasing extracellular K+ concentrations during anoxic and globally ischemic conditions (103). It appears that delayed preconditioning, irrespective of preconditioning stimulus, is always mediated by KATP channels (104). The mitochondrial KATP channel opener diazoxide significantly reduced the rate of cell death following simulated ischemia in adult ventricular cardiac myocytes (73). Intravenous injection of diazoxide ten minutes before induced ischemia greatly reduced infarct size in the rabbit heart (105); however, the protective effects of diazoxide could be blocked by 5-hydroxydecanoate (5-HD), a selective blocker of the mitochondrial KATP channel.
Making A Case For Reseveratrol As A Pharmacological Preconditioning Agent
It should be clear from the above discussion that unlike pharmacological therapeutic interventions, preconditioning protects the heart by increasing its endogenous defense mechanisms (106). Unfortunately, ischemic preconditioning–mediated cardioprotection has a limited duration––early preconditioning lasts for several hours, and delayed preconditioning lasts for several days (107). There is a definite need to identify a pharmacological preconditioning agent to render the preconditioning stimulus everlasting. Recently, monophosphoryl lipid A (MLA) was found to induce dose-dependent cardioprotection against myocardial infarction (108, 109). Such cardioprotection was achieved through the ability of MLA to increase endogenous NO formation, which participates in mediating ischemic preconditioning. Similarly, resveratrol protects the ischemic myocardium through NO (11). In this study, preconditioning of the hearts with resveratrol provided cardioprotection, as evidenced by improved postischemic ventricular functional recovery (i.e., developed pressure and aortic flow), reduced myocardial infarct size, and cardiomyocyte apoptosis. Additionally, resveratrol diminished the amount of ROS activity, which was demonstrated through reduced malonaldehyde formation. These results suggest resveratrol may act to pharmacologically precondition the heart in a NO-dependent manner.
A report by Imamura et al. showed that iNOS knockout mouse could not be preconditioned with resveratrol, further indicating that this polyphenol provides cardioprotection through NO and specifically through the induction of iNOS (51). In this study, control experiments were performed with wild type mouse hearts and with iNOS-null mouse hearts that were not treated with resveratrol. Resveratrol treated wild-type mouse hearts displayed significant improvement in post-ischemic ventricular functional recovery compared to those of non-treated wild-type hearts. Both resveratrol-treated and non-treated iNOS-knockout mouse hearts exhibited relatively poor recovery in ventricular function as compared to that in wild-type resveratrol-treated hearts. Myocardial infarct size and the number of apoptotic cardiomyocytes were lower in the resveratrol-treated wild-type mouse hearts as compared to results from the other groups of hearts. Cardioprotective effects of resveratrol were abolished when the wild-type mouse hearts were simultaneously perfused with aminoguanidine, an iNOS inhibitor. Resveratrol induced the expression of iNOS (for several hours) in the wild-type mouse hearts but not in the iNOS-knockout hearts, after only thirty minutes of reperfusion. Additionally, resveratrol-treated wild-type mouse hearts were subjected to lower amount of oxidative stress, as evidenced by reduced amount of malonaldehyde content in these hearts compared to findings in iNOS-knockout and untreated hearts. These results demonstrated that resveratrol was unable to precondition iNOS-knockout mouse hearts, but it could successfully precondition the wild-type mouse hearts, indicating an essential role of iNOS in resveratrol preconditioning of the heart. The above results strongly support the notion that resveratrol mediated cardioprotection is achieved by preconditioning (Figure 4⇓).
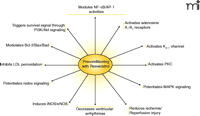
Preconditioning with resveratrol. Mechanisms of reveratrol preconditioning involve a number of well-defined molecular signaling events. The therapeutic implications of preconditioning are discussed in the text.
In normal tissue, resveratrol decreases the expression of iNOS; however, as seen from the above studies, in ischemic heart, resveratrol induces iNOS, an observation that is consistent with ischemic preconditioning (101, 108). Recently, resveratrol was found to protect the ischemic heart through the increased expression of adenosine A1 and A3 receptors (12), a property shared by ischemic preconditioning. The results of this study demonstrated significant cardioprotection with resveratrol treatment, as evidenced by improved ventricular recovery, reduced infarct size, and cardiomyocyte apoptosis. Specifically, an A1 receptor blocker (CPT; 1,3-dimethyl-8-cyclopenthylxanthine) and an adenosine A3 receptor blocker MRS 1191 [3-ethyl-5-benzyl-2-methyl-4-phenylethynyl-6-phenyl-1,4-(+/−)-dihydropyridine-3,5-dicarboxylate], but not adenosine A2A receptor blocker [CSC; 8-(3-chlorostyryl)caffeine], abrogated the cardioprotective abilities of resveratrol, suggesting a role of adenosine A1 and A3 receptors in resveratrol preconditioning. Resveratrol induced the expression of Bcl-2 and caused its phosphorylation along with phosphorylation of cyclic AMP response element binding protein (CREB), Akt, and Bad. CPT blocked the phosphorylation of Akt and Bad without affecting the phosphorylation of CREB, whereas MRS 1191 blocked phosphorylation of all these proteins. A phosphatidylinositol 3′-kinase (PI3K) inhibitor LY 294002 partially blocked the cardioprotective abilities of resveratrol (12). These results indicate that resveratrol preconditions the heart through the activation of adenosine A1 and A3 receptors, the former transmitting a survival signal through PI3K-Akt-Bcl-2 signaling pathway, while the latter protects the heart through a CREB-dependent Bcl-2 pathway in addition to the Akt-Bcl-2 pathway.
Angiogenic Properties Of Resveratrol
Therapeutic angiogenesis describes the method of improving blood flow to ischemic tissue by the induction of neovascularization by angiogenic agents administered as recombinant protein or by gene therapy. Indeed, therapeutic angiogenesis has recently emerged as a promising investigational strategy for the treatment of patients with ischemic limb and heart disease. In the past ten years, alternative revascularization/angiogenesis strategies have progressed from bench to bedside, focusing on the capillary sprouting and growth of new vessels to replace the old. However, most of the strategies involve the delivery of growth factors and, unfortunately, very little success with these strategies has been demonstrated so far for various reasons. The approach of using resveratrol to induce angiogenesis and increased expression of growth factors and their receptors is an exciting and potentially very important strategy for myocardial protection. We have shown in the rat myocardial infarction model that, three weeks after infarction, resveratrol treatment leads to increased expression of vascular endothelial growth factor (VEGF) and its tyrosine kinase receptor Flk-1 (110). Pretreatment with resveratrol also increases the amounts of iNOS and eNOS expressed as well as increases the expression of anti-apoptotic and pro-angiogenic factors NF-κB and Sp-1. We also demonstrated increased capillary density and improved left ventricular function (again, three weeks after infarction) by pharmacological preconditioning with resveratrol.
In one of our previous studies, we documented hypoxia-reoxygenation–mediated myocardial angiogenesis through an NF-κB-dependent mechanism in a rat model of chronic myocardial infarction (111). In this study, the administration of pyrrolidine dithiocarbamate (PDTC), an NF-κB inhibitor, blocked angiogenesis, thus demonstrating the essential and important role of NF-κB in myocardial angiogenesis. Again, notable induction of eNOS and iNOS by resveratrol in the myocardium provides evidence supporting the hypothesis that resveratrol regulates endothelial cell growth as also evident by increased perfused capillaries (111). The eNOS isoform is constitutively expressed and its expression can be stimulated by pharmacological interventions whereas, iNOS is a cytokine-inducible isoenzyme (112). The eNOS isoform participates in circulatory function during heart failure; the iNOS isoform may have an important role in the hemodynamics early after myocardial infarction (113). iNOS modulates arterial hemodynamics in large, conduit arteries whereas eNOS is supposed to regulate peripheral resistance of the vessel (114).
Numerous experimental results have demonstrated that very low concentrations of NO produced from eNOS or pharmacological concentrations of exogenous NO produced by NO donors reduce apoptotic cell death (115, 116). In our study, besides eNOS induction, iNOS was significantly induced in all resveratrol-treated groups. Recent studies have demonstrated that eNOS/NO play an important role in many VEGF-induced actions. VEGF induces the production of nitric oxide (NO) from rabbit, pig, bovine, and human vascular endothelial cells (117). The inhibition of NO production by eNOS inhibitors significantly reduced VEGF-induced mitogenic and angiogenic effects (118). eNOS/NO have been implicated as important mediators for VEGF-induced hemodynamic changes and microvascular permeability. Recent studies indicate that a variety of stimuli, including hypoxia, shear stress, inflammatory cytokines, high glucose, and injury can modulate eNOS expression and activity (119, 120). In vitro experiments have shown that activation of Flk-1 induces proliferation and migration of endothelial cells as well as expression of eNOS and iNOS (121, 122).
NO is a pleiotropic molecule that affects diverse biochemical and physiological function including regulation of vascular tone and vascular remodeling (123). One potential therapeutic target for NO is angiogenesis (124). Incubation of human vascular smooth muscle cells with NO donors enhances the synthesis of VEGF and inhibition of NOS abrogates VEGF production (125). Not surprisingly, eNOS inhibitors block VEGF-induced endothelial cell migration, proliferation, and tube formation in vitro and VEGF-induced angiogenesis in vivo. Dimmelers et al. found that in the absence of eNOS inhibition, VEGF stimulates PI3K- and Akt-dependent phosphorylation of eNOS, resulting in an activation of eNOS and increased NO production (126). Although both tyrosine kinase receptors Flt-1 and Flk-1 are necessary for VEGF signaling, there are differences between the intracellular functions of the two receptors (126). Flk-1, rather than Flt-1, is predominantly involved in eNOS phosphorylation, based on mutation studies. Stimulation of Flt-1 (another VEGF receptor) is linked to cell migration, whereas Flk-1 activation is associated with both cell migration and proliferation through the MAPK cascade (127). Interestingly, induction of the expression of VEGF and Flt-1 occurs within a very short time, but induction of the Flk-1 does not occur until days later (128). One of our very recent studies showed that resveratrol could induce, in chronological order, the expression of iNOS and VEGF, followed by Flk-1, and lastly eNOS. Immunohistochemistry detected increased expression of these proteins in the hearts from resveratrol-fed rats subjected to thirty minutes of ischemia and two hours of reperfusion as compared to those in non-fed hearts (129). Several other reports exist in the literature to show that resveratrol can induce eNOS and iNOS expression. For example, resveratrol induced an expression of eNOS in the human umbilical vein endothelial cells (HUVEC) (130). In addition, to its long-term effects on eNOS expression, resveratrol also enhanced the rapid production of bioactive NO, which suggests a role for iNOS. Our results support these previous observations; we have observed iNOS expression within twenty-four hours of resveratrol treatment, but eNOS expression was not apparent until three days later. Additionally, Hsieh et al. found that resveratrol induced the expression of iNOS in cultured bovine pulmonary artery endothelial cells (131).
Future Perspectives
It is becoming increasingly clear that resveratrol has two faces. On one hand, it protects cells by potentiating a survival signal. On the other hand, it selectively kills cancer cells. It behaves as an antioxidant, yet it can induce redox signaling. It is an antiproliferative agent for cancer; it induces apoptosis in tumor cells and sensitizes cancer cells by inhibiting cell survival signal transduction and antiapoptotic pathways. In contrast, the same compound triggers a survival signal in the ischemic tissue by inducing antiapoptotic genes and blocks apoptosis in ischemic heart. At low doses, resveratrol stimulates angiogenesis, but at higher doses, it blocks angiogenic response. Resveratrol has been recognized as a phytoestrogen based on its structural similarities to diethylstilbesterol (DES), yet many in vivo studies failed to confirm estrogenic potential of resveratrol. At low concentrations, resveratrol scavenges ROS, but at higher concentrations, it behaves like a prooxidant.
One likely explanation for the dichotomy of resveratrol action could arise from wide-ranging differences in tissues in pharmacokinetics, bioavailability, and metabolism. In a recent study, rats were given red wine (4 ml containing 6.5 mg/l resveratrol) and sacrificed at several time points post-ingestion (132). Resveratrol was absorbed rapidly, being detectable in plasma (18 ng/ml) and liver (20 ng/ml) within thirty minutes, in kidney (20 ng/ml) within one hour, and only detectable in heart after two hours and only at a concentration of 2 ng/ml. Thus, a low concentration of resveratrol is quite sufficient for the preconditioning of the heart. In fact, at higher concentration, resveratrol could exert toxic effect in the heart. Red wine containing 1.2 mg/l of resveratrol can inhibit platelet aggregation by 42%, even when it is diluted 100-fold (133). At a concentration as low as 100 nmol/l, resveratrol can inhibit the TNF-α-mediated expression of intracellular adhesion molecules such as ICAM-1 (134). At a concentration of 1 nM, resveratrol induces phosphorylation of ERK1/2, whereas at higher concentration of 50–100 μM, it inhibits MAPK phosphorylation (135). Interestingly, preconditioning is achieved only at a low amount of stress through the activation of MAP kinase signaling (80).
It is tempting to speculate that NO may play a crucial role in the dual behavior of resveratrol. Similar to resveratrol, NO also has two entirely opposite actions. Constitutive expression of NO is protective, but it is equally destructive to the cells at higher doses. Activation of iNOS and eNOS play a crucial role in ischemic preconditioning. It appears that resveratrol-mediated cardioprotection is achieved through the increased expression of NOS, which exerts a preconditioning-like effect, rather than direct protection.
Preconditioning, the best method yet devised for cardioprotection, is achieved by subjecting the heart to a therapeutic amount of stress, thereby disturbing normal cardiovascular homeostasis and reestablishing a modified homeostatic condition with increased cardiac defenses that can withstand subsequent stress insult. The most common and well-studied method of preconditioning is ischemic preconditioning. Such a method, however, cannot be extrapolated to clinics.
There has been a desperate search for pharmaceutical preconditioning agents. Resveratrol appears to fulfill the definition of a pharmaceutical preconditioning compound. Future studies will reveal the mystery of two faces of resveratrol and pinpoint its precise mechanism of action in cardioprotection.
Acknowledgments
This study was supported in part by NIH HL 22559, HL33889, HL56803 and HL75665, and HL 69910.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Dipak K. Das, PhD, (left) is a Professor and the Director of The Cardiovascular Research Center of the University of Connecticut School of Medicine, Farmington, Connecticut. DKD has been investigating the mechanisms of cardiac preconditioning since the late 1980s. He has pioneered work on the cardioprotective abilities of grapes and wine and their components, including resveratrol. His work has established that resveratrol provides cardioprotection by pharmacologically preconditioning the heart.
Nilanjana Maulik, PhD, FACN, FICA, (right) is an Associate Professor of Department of Surgery, University of Connecticut Medical Center, Farmington, Connecticut. NM has been studying the cellular and molecular mechanisms underlying the delayed protective effects of ischemia- and pharmacologic-induced preconditioning against myocardial infarction. Her work recently established that resveratrol at low dose is pro-angiogenic, which leads to cardioprotection.

