Disrupting Calcium Channel Expression To Lower Blood Pressure: New Targeting of a Well-Known Channel
Voltage-gated calcium channels in vascular smooth muscle cells (VSMCs) mediate many crucial Ca2+-dependent processes inherent to vascular biology including arterial contraction and gene expression. As such, they are intricately involved in the regulation of blood flow to critical organs and the determination of blood pressure levels. The VSMCs lack action potentials that form the basis for excitability in neuronal and cardiac cells. Instead, under physiological conditions, VSMCs show steady-state resting membrane potentials that fluctuate between −60 mV and −40 mV in response to vasoactive substances. Within this voltage range, membrane depolarization and the open-state probability of Ca2+ channels are positively and tightly coupled to regulate Ca2+ influx and vascular tone (1). Mathematical modeling suggests that at least several thousand pore-forming Ca2+ channel subunits are expressed in the plasma membrane of a single VSMC (2). The channels open but only slowly inactivate during sustained depolarization, so that Ca2+ influx mediated by only a fraction of the channel population appears sufficient for tonic vasoconstriction.
Although the voltage-dependent Ca2+ channels in VSMCs play a central role in excitation-contraction coupling in small arteries, the identity of their molecular composition is largely unknown. Similarly, there is little knowledge about the physiological factors that regulate their expression under physiological conditions and during disease states. This knowledge gap may impair our ability to formulate new therapeutic interventions for the treatment of common vascular diseases that involve Ca2+ channel abnormalities. For example, recent findings suggest that abnormalities of vascular Ca2+ channels may contribute to the development of hypertension, a condition that currently affects more than 40 million Americans (3). Thus, healthcare concerns and costs dictate the importance of identifying the structural proteins that compose the Ca2+ channels in VSMCs, and suggest the need to define the factors that influence their expression and function.
In this regard, studies in other cell types have firmly established that voltage-dependent Ca2+ channels are multi-protein complexes composed of a pore-forming α 1 subunit and accessory β and α 2δ subunits (Figure 1⇓) (4–9). The α 1 subunit confers signature properties to the channel including voltage sensing, Ca2+ permeability, and inhibition by Ca2+ channel blockers. The structure of the α 1 subunit includes four repeat domains (I, II, III, and IV) each composed of six transmembrane segments (10, 11). Signaling molecules, such as protein kinase A (PKA) and protein kinase C (PKC), which are important regulators of vascular tone, can bind to intracellular domains to modify the gating of the Ca2+ channel. Additionally, the N- and C-terminal regions of channel subunits can modulate channel behavior. For example, the C terminus constitutively binds calmodulin to permit Ca2+-dependent inactivation of the channel (12–17).
Importantly, although ten genes are available to encode different α 1 subunits, a single CaV1.2 gene appears primarily to encode the voltage-gated Ca2+ channels in VSMCs that show high-threshold, long-lasting (i.e., “L-type”) Ca2+ currents and that are sensitive to inhibition by 1,4-dihydropyridine antagonists. Indeed, the vascular CaV1.2 (α 1C) gene represents a smooth muscle splice variant (α 1C-b) that differs in four regions from the splice variant (α 1C-a) expressed by cardiac myocytes (18, 19). Alternative splicing of exon 8 results in amino acid differences in transmembrane segment IS6 that confer an increased sensitivity for 1,4-dihydropyridine drugs (18, 19). Further alternative splicing of exon 1 results in a shortened N terminus that may permit a separate promoter to regulate selectively the expression of CaV1.2 channels in VSMCs without altering channel availability in other cell types (20, 21). Not surprisingly, the α 1C subunit plays a key role in mediating vascular reactivity. For example, depolarization-induced contraction and myogenic tone are abolished in the arteries of mice showing smooth muscle-specific inactivation of the CaV1.2 (α 1C) gene. In these animals, the average systemic blood pressure was ~30 mm Hg lower than in control mice, reflecting the key role of CaV1.2 channels in mediating resting vascular tone and normal levels of blood pressure in vivo (22).
Although the α 1 subunit represents the central protein that forms the pore of voltage-gated Ca2+ channels, other subunits are necessary for full physiological function. As studied in most detail in neurons and cardiac myocytes, four different genes encode populations of CaVβ subunits (β 1, β 2, β 3, and β 4) that are expressed in a tissue-specific manner (10, 23–25). Although heterogeneous populations of β subunits appear to be expressed in distinct cell types, a single β subunit may critically determine the availability of functional CaV1.2 channels. For example, conventional β 1-null mice die from asphyxia at birth, revealing the requisite contribution of the β 1 subunit to Ca2+ channel expression in skeletal muscle cells (26). In contrast, β 2-null mice die of a cardiac defect in utero (27) and β 4-deficient mice show primarily neural deficits that include a “lethargic” and “stargazing” phenotype (28). Importantly, all of the β subunits belong to the membrane-associated guanylate kinase (MAGUK) family of scaffolding proteins (29), which possess a guanylate kinase (GK) and a Src homology 3 (SH3) domain (Figure 1⇓) (30, 31). The GK region of the β subunit contains a beta interactive domain (BID) that is required for high affinity interaction with the alpha interactive domain (AID) on the intracellular I–II linker of the α 1 subunit (32–35). Other lower affinity interactive domains also have been reported (10, 35, 36). Importantly, all four β subunits modulate the availability of α 1 subunits at the surface membrane and influence channel properties in an isoform specific manner. As scaffolding proteins, the β subunits are thought to localize and integrate intracellular signals to influence channel behavior and drug sensitivity (30, 37–39). In addition to the β subunits, four distinct α 2δ subunits that arise from alternative splicing of a single gene also may interact with the α 1 subunit to influence channel expression and properties (40, 41).
The identities of the β subunits and α 2δ subunits that contribute to the formation of functional CaV1.2 channels in VSMCs are largely unknown. Based on the detection of many of the corresponding genes, however, a diverse composition of CaV1.2 vascular channels is anticipated [for review, see (42)]. For example, transcripts encoding all four β subunits (β 1, β 2, β 3, and β 4) have been detected in VSMCs from different arteries, although only expression levels of the β 2 and β 3 proteins have been confirmed. Similarly, at least two α 2δ genes have been detected. Notably, the impact of β gene deletions on the expression of vascular CaV1.2 channels has only been examined in β 3 null mice (43). In these animals, the density of voltage-gated Ca2+ current in aortic VSMCs was reduced by 30% compared to cells from control animals. However, the β 3 null mice showed normal levels of blood pressure, suggesting that the loss of Ca2+ current in the aortic myocytes did not extend to the VSMCs of the resistance vessels involved in blood pressure regulation. Alternatively, the activation of compensatory mechanisms may have restored blood pressure to normal.
Although the precise composition of CaV1.2 channels in VSMCs awaits definition, an upregulation of these channels is regarded as part of the extensive biological and morphological adaptations that characterize the vasculature during the pathogenesis of hypertension (42, 44). The initial rise in blood pressure that signals the onset of genetic or “essential” hypertension likely originates from the interaction between multiple predisposing genes and environmental factors. However, if blood pressure is not restored to normal levels by compensatory mechanisms, the ion channels in the VSMCs of the arterial circulation appear to “electrically remodel” to sustain arterial contraction (44). A central component of this remodeling includes the rapid and persistent upregulation of functional CaV1.2 channels in the affected VSMCs (45–50). Importantly, increased expression of vascular CaV1.2 channels has been reported in rats with genetic and renal forms of hypertension (47, 50), suggesting that this event is not limited to single neural or endocrine profiles of the disease. For example, the mesenteric arteries of spontaneously hypertensive rats (SHR) with a genetic form of hypertension show an overabundance of CaV1.2 channels compared to similar arteries from the control Wistar Kyoto (WKY) rat strain (50). This abnormality has been shown to extend to the renal and skeletal muscle circulations of the SHR and corresponds to a higher density of Ca2+ current in the affected VSMCs and an accentuated Ca2+-dependent tone in arteries isolated from these vascular beds (45, 47, 50–53). Interestingly, a recent report suggests that even mild and short-term increases in blood pressure are sufficient to trigger the upregulation of CaV1.2 channels in small arteries in vivo, inferring that this abnormality may occur early in the pathogenesis of hypertension (47).
Notably, an increased availability of functional CaV1.2 channel protein, rather than altered properties of the channel, appears to fully account for the elevated voltage-gated Ca2+ current in the VSMCs of hypertensive animals. Even early in the development of genetic hypertension, the elevated Ca2+ current in VSMCs does not relate to an altered voltage-sensitivity of the channel (45, 52), and normal properties of the channel persist during the chronic stage of hypertension (47). Also, single-channel recordings detect an increased number of Ca2+ channel openings without evidence of altered single-channel conductance or open-time distribution (53). All of these findings are consistent with the hypothesis that an increased availability of characteristic CaV1.2 channels is the fundamental abnormality in the VSMCs of hypertensive animals.
The stimuli that link the development of high blood pressure and CaV1.2 channel expression remain unclear. At least two reports have suggested that membrane depolarization, a characteristic feature of VSMCs exposed to high blood pressure, may dynamically increase the expression of vascular CaV1.2 channels (47, 54). Small increases in CaV1.2 mRNA have been detected in vessels from hypertensive rats, but these do not appear to fully account for the striking over-abundance of the CaV1.2 protein (50, 55). For this reason, it is likely that events that enhance CaV1.2 channel translation, translocation, or stability in the plasma membrane represent the primary abnormal processes.
Despite the recognition that vascular CaV1.2 channels upregulate during hypertension, antihypertensive therapy directed at CaV1.2 channels is still limited to blocking Ca2+ influx through these channels using traditional calcium channel blockers (CCBs) (56, 57). In the United States, three classes of CCBs are prescribed, including the phenylalkylamines, the benzothiazepines and the dihydropyridines. These drug classes bind to distinct but closely related sites on the CaV1.2 channel to reduce the open-state probability of the CaV1.2 channel and thereby attenuate the voltage-gated Ca2+ influx that is required for vascular activation. Interestingly, the increased abundance of the vascular CaV1.2 channels during hypertension may provide more binding sites for the CCBs, possibly explaining why their antihypertensive effects are more pronounced in hypertensive compared to normotensive animals and humans (56, 58–60). Regardless, the antihypertensive effect of the CCBs and the risk of cardiovascular events varies between patient populations (61), suggesting that in addition to improved drug profiles, novel interventions for reducing Ca2+-dependent tone may improve patient outcomes. In this respect, using molecular interventions to reverse the abnormal expression of CaV1.2 channels rather than attempting to block Ca2+ influx, may represent a helpful approach.
Two strategies for blunting Ca2+ channel expression have been achieved experimentally. Both rely on altering the assembly and release of CaV1.2 channels from the endoplasmic reticulum (ER). Notably, normal assembly and trafficking of CaV1.2 channels relies on the co-assembly of α-β complexes in the ER, which is mediated by a high affinity interaction between the AID and BID of the α and β subunits, respectively. This α-β interaction masks ER retention sequences on the intracellular I–II linker of α 1C, thus enabling translocation of the α-β channel complex from the ER–Golgi complex to the plasma membrane (Figure 2A⇓) (62, 63). One tactic for reducing the number of functional CaV1.2 channels involves the introduction of mutated β subunits that co-assemble in the ER with the α 1C subunit, but prevent its translocation to the plasma membrane (64, 65). These β subunit “decoys” retain an intact BID and bind with high affinity to the AID of α 1C but lack the ability to mask the ER retention sites on the α 1C subunit to enable transit from the ER–Golgi complex. As a result, the decoys form α 1C-β decoy complexes that remain sequestered in the ER. Concurrently, the number of functional CaV1.2 channels at the plasma membrane is reduced (Figure 2B⇓). Proof of principle has been demonstrated in cardiac myocytes in which overexpression of BID with truncated or missing N- or C-terminal regions decreased the plasma membrane localization of α 1C and also reduced voltage-gated Ca2+ current and contractile function (66, 67). A similar loss of functional CaV1.2 channels was achieved by the adenoviral introduction of β subunits with truncated N- and C-terminal regions (67). Notably, the β decoys reduced the excitation-contraction coupling that relies on Ca2+ entry through CaV1.2 channels, but did not disrupt the kinetics of contraction or relaxation of the cardiac cells (66).
A second molecular strategy for disrupting the expression of CaV1.2 channels involves overexpressing “lure sequences” that include the AID of the α 1C subunit (34, 68). Using lure sequences as baits for BID by overexpressing the AID-containing I–II linker of the α subunit, the availability of endogenous β subunits to bind to the AID of α subunits and permit the trafficking of α-β complexes to the plasma membrane is reduced (Figure 2C⇓) (68). Proof of principle for this approach has been demonstrated for CaV2.1 channels in neurons but, presumably, could be used to ameliorate CaV1.2 channel expression in VSMCs. Notably, the use of both of these molecular strategies to disrupt CaV1.2 channel assembly and trafficking would require VSMC-specific tissue targeting. To accomplish this goal, a SM22α promoter that regulates smooth muscle cell–specific gene expression has been described that drives the functional expression of genes in small arteries in vivo (69, 70). Viral vectors are being designed that incorporate these promoter elements with specificity for smooth muscle cells, thereby limiting the overexpression of therapeutic molecules designed to downregulate CaV1.2 channels to the vasculature. Additionally, alternative promoters for the vascular and cardiac forms of the α 1C subunit were recently identified, and analysis of the 5′ flanking sequence for these genes has revealed positive and negative regulatory elements (64, 71–73). Based on these findings, an emerging strategy for selectively decreasing CaV1.2 channel expression in VSMCs may include diminishing the expression of the vascular isoform of CaV1.2 channels by enhancing the level of negative regulatory transcription factors.
It is possible that molecular interventions to reverse the over-expression of vascular CaV1.2 channels in hypertension may have some advantages over traditional CCB therapies. First, even the long-acting CCBs require daily administration, whereas new therapeutics that use viral vectors to target the vascular-specific expression of CaV1.2 channels may have more enduring effects. Second, although newer CCBs show improved clinical profiles including higher degrees of vascular selectivity and fewer cardiac and renal side effects (74–77), genetic-based approaches may offer even higher vascular selectivity. For example, the presence of a vascular splice variant of the α 1C subunit (18, 19) affords a tantalizing opportunity to differentially downregulate the CaV1.2 channels in the vasculature to reduce blood pressure while maintaining expression in the heart, which relies on CaV1.2 channels for cardiac contraction.
Overall, the vision underlying these novel approaches to treat hypertension predicts that a direct attack on the pathophysiology of the disease by reducing the over-abundance of functional CaV1.2 channels has potential therapeutic advantages as new or adjunct therapy to lower blood pressure. Although simple in concept, the optimization and translation of these molecular therapies will require a detailed knowledge of the mechanisms that regulate CaV1.2 channel assembly and trafficking in VSMCs, and the events that trigger their pathogenic upregulation during the development of hypertension.
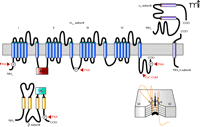
Proposed topology of the CaV1.2 channel. The formation of the pore by the α 1C subunit is shown (lower right). Genes for four β subunits (β 1, β 2, β 3, and β 4) and four splice variants of α 2δ subunits have been identified. The β subunit contains a beta interactive domain (BID), which is required for high affinity interaction with the alpha interactive domain (AID) of the α subunit. The subunits also have phosphorylation sites and binding sites for signaling molecules. The formation of the pore by the α 1C subunit is shown (lower right). CaM, calmodulin; PKA, protein kinase A; PKC, protein kinase C.
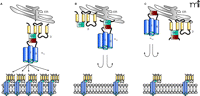
Molecular interventions to reduce the expression of functional CaV1.2 channels. A. The co-assembly of α-β complexes in the endoplasmic reticulum (ER) permits translocation of CaV1.2 channels to the plasma membrane to form functional channels. B. Overexpressed mutant β subunits co-assemble in the ER with the α 1C subunit but prevent its translocation to the plasma membrane. These “decoy” β subunits retain the BID that permits interaction with the AID of the α subunit, but lack the ability to mask the ER retention signal to enable exit of the α-β complex from the ER. C. Overexpression of the AID of the α 1C subunit acts as bait for the BID of endogenous β subunits, thereby reducing their availability to bind to endogenous α 1C to permit its exit from the ER.
Acknowledgments
Supported by USPHS R01 HL-59238 and HL-68406 from the National Institutes of Health
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Nancy Rusch, PhD, is Professor and Chair of the Department of Pharmacology and Toxicology at the University of Arkansas for Medical Sciences. She made some of the initial observations about calcium and potassium channel dysregulation in small arteries during the development of hypertension. She continues to explore mechanisms that induce vascular ion channel abnormalities in vasospastic diseases. Please direct correspondence regarding this article to NR. E-mail: nrusch{at}uams.edu; fax 501-686-5521.

James Marsh, MD, is Professor and Chair of the Department of Internal Medicine at the University of Arkansas for Medical Sciences. He has focused for many years on the problem of understanding how a cardiac myocyte or vascular smooth muscle cell determines how many calcium channels to express on the cell membrane under physiological and pathophysiological conditions. He has made significant discoveries about transcriptional regulation of calcium channel genes in response to neurohormonal stimuli. An active cardiologist, he deals with problems of ion channel dysregulation in the clinic as well as in cell biology. E-mail: jmarsh{at}uams.edu.

Mony Fraer, MD, is an Assistant Professor of the Department of Internal Medicine at the University of Arkansas for Medical Sciences. He is board certified in Internal Medicine and Nephrology, and is an active clinician and clinical educator. His special areas of interest include resistant hypertension, bone and mineral metabolism, and acid-base and electrolyte abnormalities. E-mail: mfraer{at}uams.edu.

Swapnil Sonkusare, MS, is a graduate student in the Department of Pharmacology & Toxicology at the University of Arkansas for Medical Sciences. He is performing research to pinpoint the contributions of distinct β subunits to vascular calcium channel expression, and devise molecular methods to interrupt the interactions of the subunits. E-mail: ssonkusare{at}uams.edu.

