Nicotinic and Opioid Receptor Interactions in Nicotine Addiction
Most addictive drugs affect behavior primarily through interactions with specific membrane receptors, which alter neuronal activity. The effects on neurons, combined with complex behavioral, contextual, and social interactions, induce long term changes in brain activity that underlie drug dependence (1–4). The precise molecular events that characterize these long term changes are the subject of intense investigations within the field of drug abuse research. An important focus of these efforts is the mesoaccumbens dopamine system.
Ventral Tegmental Area (VTA) dopamine neurons send projections to many brain areas, including the nucleus accumbens (NAcc), prefrontal cortex (PFC), and basolateral amygdala (BLA). Dopamine release from these neurons has been correlated with the rewarding effects of many addictive drugs, including nicotine and opiates. Within this system, nicotine activates nicotinic acetylcholine receptors (nAChRs), whereas opiate drugs primarily activate mu opioid receptors (MOR). These receptor classes are quite distinct in their structure and function, but the endpoint of activating each of these classes has striking similarities with regard to changes in neuronal excitability.
Nicotinic receptors are composed of five protein subunits that surround a central ion channel. There are twelve different subunit genes that are expressed in the vertebrate nervous system, and the different gene products can combine in a variety of ways to form functional receptors (5, 6). Within the dopamine system, mRNAs encoding α2-α7 and β2-β4 are expressed, leading to considerable diversity in nAChR subtypes (7, 8). Pharmacological approaches, sometimes in combination with genetic deletion of specific nAChR subunits in mice, have provided important insights into the subunits that contribute to VTA dopamine neuron excitability and reward-associated behavior (7–14). Unfortunately, these observations suggest that there is not a single subunit or subunit combination that underlies nicotine addiction, although we are getting closer to identifying the important subtypes. The extent to which these receptors may be molecular targets for therapeutics is a subject of debate and intense investigation, with the two main hurdles being subtype identification and specificity for this pathway.
Results from cellular and synaptic assays of nAChR function within the VTA suggest that α7 homomeric receptors are expressed by dopamine neurons and on presynaptic glutamatergic inputs to these cells (11, 12). Thus, α7* nAChRs (where * indicates the possible contribution of other subunits to these receptors) are positioned to increase neuronal excitability pre- and postsynaptically. Indeed, activation of presynaptic α7 receptors with low, physiologically relevant nicotine concentrations contributes to long-term potentiation (LTP) at the excitatory inputs to VTA dopamine neurons (15). Both dopamine and non-dopamine neurons within the VTA also express non-α7-containing nAChRs and the makeup of these receptors varies with cell type. γ-Amino butyric acid (GABA)ergic interneurons likely express α4β2* receptors, although other subunits may contribute to the α4β2* receptors and/or other pentameric combinations within local inhibitory circuitry of the VTA (7, 8, 11–16). DA neurons also express α4β2 nAChRs with important contributions of α6, α5, and β3 (7, 8, 13, 14). The non-α7-containing nAChRs generally have a high affinity for nicotine and have been implicated in the direct excitation associated with arrival of the drug. With prolonged nicotine exposure, non-α7-containing nAChRs can be “upregulated,” which is a phenomenon associated with increased binding of radiolabeled ligands and stronger functional responses (17–20). The impact of nAChR upregulation on dopaminergic signaling is an area of active ongoing investigations.
The three major subtypes of opioid receptors––mu, delta, and kappa (MOR, DOR, and KOR, respectively)––were originally identified pharmacologically and more recently through molecular cloning (21). Drugs that activate MORs, DORs, or both, are generally reinforcing when tested in animals or human subjects, whereas the activation of KORs leads to aversive behavioral effects, which are described as anhedonic (21). These different behavioral effects are mediated, in part, by differential expression of these receptors on different neuronal types within the mesoaccumbens system (21). Using fluorescent labeling of VTA neurons that project to NAcc or to BLA, Ford et al. demonstrated that there is considerable diversity in the physiology of these cells, depending upon their postsynaptic targets. In that context, the authors found that MOR and DOR receptors caused a weak inhibition of VTA neurons that project to the BLA and had no effect on neurons projecting to NAcc (22). The KOR responses had the opposite pattern with strong inhibition of the NAcc-projecting neurons and a relatively smaller inhibition of the BLA projections (22). The diversity of the VTA dopamine neurons also extended to the modulation of afferent inputs. Release of GABA within the VTA is acutely inhibited by opioids and is chronically altered during withdrawal from chronic opioid use (21). As with direct opioid inhibition, the VTA neurons with different projections showed differential modulation of GABAergic inputs in response to KOR and MOR/DOR activation.
VTA dopamine neurons are known to release dopamine within the cell body region as well as from the axonal projections. The opioid effects outlined above impact dopamine release in both regions. Dopamine neurons express D2-type dopamine receptors that mediate feedback autoinhibition through inhibitory postsynaptic currents (IPSCs) (23, 24). Activation of MORs or DORs had no effect on the D2-mediated IPSCs measured on dopamine cells in all areas. Consistent with the large inhibitory somatic responses outlined above, KOR activation caused stronger inhibition of the NAcc-projecting VTA dopamine neurons (22). This differential regulation of specific pathways via opioid receptor activation highlights the fact that the dopamine neurons are a heterogeneous population, with target tissue–dependent expression of specific receptor classes. These differences certainly contribute to the behavioral effects of opiate drugs and provide insight into potential therapeutic targets for treatment of drug addiction.
At the sites of dopamine release in the NAcc, nicotinic and opioid receptor ligands exhibit profound effects. In addition to the potent effects of systemic injections of nicotine and MOR agonists on dopamine release, enhanced dopamine release can also be seen with administration of these compounds directly in the NAcc (25–28). Systemic administration modulates the dopamine neurons (directly) and their afferent inputs, causing the dopamine neurons to transition from tonic to burst firing patterns (11, 14–16, 21, 29–31). However, the mechanisms underlying the enhancement of DA release following injections of these drugs into the NAcc is less clear.
Activation of MORs and DORs expressed on cholinergic inter-neurons within the NAcc, inhibits ACh release from these neurons, lowering extracellular ACh levels (32, 33). Thus, a straightforward hypothesis is that a reduction in AChR activity in the NAcc promotes DA release (33–35). But a conflict exists between this hypothesis and synaptosomal evidence demonstrating that activation of presynaptic nAChRs on DA terminals triggers transmitter release (36). Recent electrochemical measurements of evoked DA release in tissue slices provide some resolution, as they demonstrate that nAChR agonists or antagonists suppress DA release by either desensitizing or by blocking the effects of endogenous ACh on the DA terminals (Figure 1⇓). Interestingly, this suppression can be overcome by high frequency burst stimuli leading to even stronger DA release (37–39). These findings suggest that in concentrations relevant to cigarette smoking, nicotine only transiently activates nAChRs on DA terminals before strongly desensitizing them. This blockade of nAChR activity removes the influence of endogenous ACh, thus leading to a high, basal probability of release from DA terminals. This change simultaneously sensitizes DA terminals to burst firing patterns through a process that is not entirely clear.
Thus, MOR and DOR agonist-induced enhancement of dopamine release following focal NAcc administration is mediated by a modulation of local cholinergic tone. In contrast, KOR agonists administered into the NAcc only inhibit local dopamine release and this is likely mediated by KORs expressed directly on the dopamine terminals (40). The effects of the various opioid receptors on the frequency-dependence of dopamine release remains to be tested. Clearly, the cellular distribution of these various receptor classes allows for differential modulation of DA release by nicotinic and opioid receptor systems and provides insight into the NAcc microcircuitry.
Several lines of evidence suggest profound interactions between the nicotinic and opioid receptor systems, particularly in the context of nicotine addiction. Acute administration of nicotine in vivo increases the release of endogenous opioids (41, 42), and chronic nicotine treatment increases expression of MORs in striatum of female rats (43) and in VTA of mice (both sexes) (44). Numerous behavioral studies have implicated opioid receptors in the rewarding effects of nicotine and an increased MOR expression could be a contributing factor. For example, the opioid receptor antagonist nalox-one attenuates nicotine-induced increases in behavioral responses to food reward (45) as well as withdrawal symptoms in nicotine-dependent animals (46). In human opiate drug users there is an exceptionally high prevalence of smoking (47, 48). Also in human smokers, the likelihood that individuals will remain abstinent at the end of smoking cessation treatment has been linked to a single missense nucleotide polymorphism (SNP) in exon 1 [Asn40Asp (A118G)] of the mu opioid receptor gene, OPRM1 (49). Interestingly, the most profound association of the OPRM1 variant with the relative reinforcing value of nicotine was found in female test subjects (50), mirroring the preclinical studies in rats cited above (40). It is important to note that many rodent studies at the behavioral and cellular levels have not considered sex differences as a potential contributing factor to experimental outcomes.
Using transgenic animals Blendy and colleagues have examined the relationship between nicotine’s behavioral effects and MOR expression (44). The nicotine-mediated activation of the transcription factor cAMP-responsive element binding protein (CREB) is required for the rewarding effects of the drug, as assessed by a conditioned-place preference behavioral screen. This behavioral assay tests not only the drug effects but also the influence of contextual cues, which are a critical element in the rewarding effects of most drugs of abuse, including nicotine (51). Pharmacological or genetic disruption of MOR function in these mice interfered with nicotine-conditioned place preference, and the nicotine-induced changes in MOR expression were found to be CREB dependent. Together their findings suggest that CREB-dependent increased expression of functional MORs is required for nicotine-conditioned reward.
The persistent behavioral effects of nicotine arise from numerous cellular and molecular alterations in the smoker’s brain. Pharmacological and molecular assays have identified nicotinic and opioid receptor classes expressed within the mesoaccumbens dopamine system and these are clearly involved in the rewarding effects of nicotine and opiate drugs. We have only begun to explore the interactions between these receptor systems that may be critical for the development of addiction to nicotine and possibly other drugs of abuse. With luck, new treatment strategies may arise from these efforts to help treat those individual who are habitual users of nicotine and opiates.
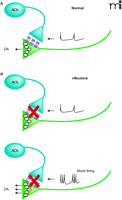
A diagram of the modification of dopamine release dynamics by nAChRs on dopamine terminals. A. The illustration indicates control conditions, where endogenous ACh release from cholinergic interneurons causes a high probability of dopamine release. Low frequency stimulation results in dopamine release that is very similar to that seen with high frequency burst-type stimulation. B. Nicotine treatment (upper panel) desensitizes the presynaptic nAChRs on the dopamine terminals, lowering release probability and decreasing the dopamine secretion elicited by low frequency stimulation. Under these conditions, high frequency burst firing (lower panel) causes more dopamine release than that seen under control conditions. Similar effects are seen with nAChR antagonists.
Acknowledgments
I would like to thank J.P. Britt, J.T. Williams, J. Blendy, and C. Lerman for helpful comments on this manuscript. This work was supported by a grant from NIDA DA015918 to DSM.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Daniel S. McGehee, PhD, is an Associate Professor in the Department of Anesthesia & Critical Care at The University of Chicago, and is a member of Committee on Neurobiology, and the Committee on Cell Physiology. His laboratory studies the cellular and synaptic mechanisms underlying nicotine addiction. He is also investigating the modulation of pain by nicotinic receptors. E-mail: dmcgehee{at}uchicago.edu; fax: 773-702-4791.

