G Protein-Coupled Time Travel: Evolutionary Aspects of GPCR Research
- Holger Römpler1,3,
- Claudia Stäubert1,
- Doreen Thor1,
- Angela Schulz1,
- Michael Hofreiter2 and
- Torsten Schöneberg1
- 1Institute of Biochemistry, Molecular Biochemistry, Medical Faculty, University of Leipzig, Johannisallee 30, 04103 Leipzig, Germany.
- 2Department of Evolutionary Genetics, Max Planck Institute for Evolutionary Anthropology, Leipzig, Germany.
- 3Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, USA
Abstract
The common seven-transmembrane–domain (TMD) architecture of G protein–coupled receptors (GPCRs) has been preserved over a vast period of time, and highly conserved amino acid motifs and residues have evolved to establish ligand and signal transduction specificities. The mining of evolutionary data from sequenced genomes and targeted retrieved orthologs has proven helpful for understanding the physiological relevance of individual GPCRs and for interpreting the clinical significance of GPCR mutations in structural terms. Sequence analysis of GPCR pseudogenes, which are considered as genomic traces of past functions, as well as recent success in sequence analysis of GPCR genes from extinct species, provide further information. This review discusses recent advances and approaches aimed at developing a better understanding of GPCR biology based on evolutionary data.

Introduction
G protein–coupled receptors (GPCRs) constitute the most prominent family of validated pharmacological targets in biomedicine. Approximately sixty percent of approved drugs elicit their therapeutic effects by selectively targeting members of this receptor family (1). Despite the remarkable structural diversity of natural GPCR agonists and low sequence homology among GPCRs, hydropathy analysis and biochemical data suggest that all GPCRs share a common molecular architecture consisting of seven transmembrane domains (TMDs) connected by three intracellular and three extracellular loops. Currently, a high-resolution structure is available only for bovine rhodopsin (2, 3), and so broader consideration of GPCRs must rely on other sources of structural information. The availability of in-depth genome sequence data for human (4), mouse (5), and many other vertebrates, as well as of numerous invertebrates, has provided the unique opportunity to compare and analyze genome-wide GPCR sequences. Such comparative genomic approaches can provide information about the origin and evolutionary history of GPCRs, illustrated below in the first two paragraphs by the example of rhodopsin-like GPCR. Comparative genomic data provide information about the origin and evolutionary history of GPCRs, and analyses of genomes have revealed hundreds of obviously non-functional gene-like sequences, so-called pseudogenes, many of which reflect GPCR-like sequences suggestive of past functions. Finally, comparative genomic approaches can help to identify conserved sequence motifs that are responsible for certain aspects of GPCR functionality, which, through mutation, may cause disease.
The Origin of Modern GPCRs
The evolutionary success of the GPCR superfamily is reflected both by its presence in almost every eukaryotic organism and by its abundance in mammals. Based on the genome sequences of humans and other mammalian species, approximately three to four percent of mammalian genes code for GPCRs. The GPCR superfamily comprises at least five structurally distinct families: the Glutamate, Rhodopsin, Adhesion, Frizzled/Taste2, and Secretin (GRAFS) families (6, 7). The rhodopsin-like receptors (also called family A or 1) form the largest family in vertebrates. Because there is very little sequence homology among the rhodopsin-like, secretin-like (family B or 2), and glutamate-like (family C or 3) receptor families, the evolutionary origin of GPCRs and their ancestry remain a matter of debate.
Proteins that display a seven-TMD architecture are present in prokaryotes that utilize light-sensitive proteo-, halo- and bacteriorhodopsins to harvest energy and fix carbon via a non-chlorophyll-based pathway. In addition to the ion-translocating rhodopsins, sensory rhodopsins exist in Halobacteria that promote phototaxis by collaborating with transducer membrane-embedded proteins with no relation to G proteins [i.e., halobacterial transducer of sensory rhodopsin-(Htr)-I and -II]. Like the rhodopsins of bilateral animals, prokaryotic rhodopsins contain retinal covalently bound to TMD7. Similarly, eukaryotes contain transmembrane proteins that are structurally similar to prokaryotic sensory rhodopsins (8, 9). The structural and functional features shared by prokaryiotic and eukaryotic rhodopsins suggest a common ancestry; however, despite these similarities, sequence comparisons provide no convincing evidence of an evolutionary linkage between prokaryotic rhodopsins and eukaryotic GPCRs (10).
Signal transduction through G proteins is definitive of GPCRs. The rhodopsins of insects and vertebrates are coupled to the G proteins Gq and transducin, respectively. Several groups of worms (e.g., nematodes and trematodes) are the evolutionarily oldest extant lineages that exhibit evidence of rhodopsin- and G protein–mediated photoreception (11). These appeared, however, long after GPCRs and G protein signaling evolved. Structural and functional data clearly show that G protein signaling via GPCRs is present in yeast/ fungi (12) and plants (13) (e.g., fungal pheromone receptors, slime mold cAMP receptor–like receptors, and glutamate receptor–like receptors), as well as in unicellular eukaryotes, such as the slime mold Dictyostelium discoideum (14). GPCRs with some structural relations to cAMP receptor–like receptors and adhesion receptors are also found in plant genomes. Glutamate receptor–like receptors are present in D. discoideum and the sponge Geodia cydonium (15), which diverged more than 600 million years ago. Sponges (Porifera) belong to the earliest occurring metazoans (animals). In contrast, the structural signatures of rhodopsin-like GPCRs do not appear to extend phylogenetically back before the appearance (approximately 580–800 million years ago) of Bilateria such as insects, mollusks, nematodes, and trematodes (16–18). Given that the coupling of G proteins to seven-TMD proteins evolved before the plant/fungi/ animal split (i.e., about 1.2 billion years ago), and that the first rhodopsin-like receptors appeared early in metazoan evolution (Figure 1⇓), the retinal-based photosensory system likely represents a “re-invention” (i.e., a product of convergent evolution). Among the rhodopsin-like GPCRs, serotonin receptors appear to be among the oldest, as suggested by their presence in planarians (19) and in many nematodes (20).
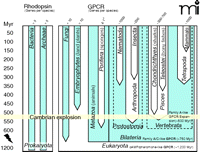
Representation of GPCRs in major evolutionary lineages. Heptahelical transmembrane-spanning proteins of the proteo-, halo-, and bacteriorhodopsin families are present in prokaryotes. Except for their transmembrane architecture, there is no evidence of an evolutionary relation between the genuine GPCRs and bacteriorhodopsins, which are not coupled to G proteins. GPCRs and their signal transduction probably evolved ~1.2 billion years ago. Genomes of extant plants and fungi usually contain less than 10 GPCR genes. The first rhodopsin-like GPCRs, which compose the main GPCR family in vertebrates, appeared ~580–800 million years (Myr) ago. Expansion of rhodopsin-like GPCRs started ~500 Myr ago, giving rise to over 1000 members in mammalian genomes. The relationships of the major lineages, which remain controversial, are not indicated.
a Complete genome data are not yet available.
The Evolutionary Expansion of Rhodopsin-like GPCRs
Vertebrates appeared in evolution about 500 million years ago, during the Cambrian period. With the exception of odorant receptors (ORs), rhodopsin-like receptors occur in vertebrate genomes twice as frequently as they do in invertebrates (Figure 1⇑). Although they are the predominant family of GPCRs in vertebrates as a whole, rhodopsin-like receptors occur significantly less frequently in more basal vertebrate lineages than in higher species, which primarily reflects the continuous expansion of the OR gene family (7). The evolutionary rise in the number of GPCR genes is generally explicable in terms of two mechanisms of duplication, described below. Subsequent to gene duplication, new functions can evolve as one or even both members of a duplicated gene pair mutate and acquire novel functionality; the fitness of the organism is presumptively less at risk to mutation in the context of multiple gene copies. Disadvantageous mutations in GPCRs are removed from a population through purifying selection, and many evolutionarily old GPCR genes, including the rhodopsins, display strong purifying selection (21–23).
Intrachromosomal Gene Duplications
Clusters of GPCR genes occur as the result of contiguous duplication of genes. The most impressive example is found in ORs; human chromosome 11 encodes nearly half of the classical OR repertoire, including a single cluster of more than 100 OR genes (24). Multiple copies of related GPCR genes, such as the human protease-activated receptors (human 5q13), P2Y12-like receptors (human 3q24), trace amine associated receptors (TAAR; human 6q23.2) and CC-chemokine receptors (human 3p21.3), are arranged in a tandem-like fashion.
Entire Chromosomal and Whole Genome Duplications
The dopaminergic receptor paralogs DRD5 and DRD1, and the adrenergic receptor paralogs ADRA2C and ADRA1 on chromosomes 4 and 5, respectively, seem to have originated from a chromosomal or even a whole genome duplication event (25–28). Polyploidy is common in certain vertebrates, such as fish and amphibian species; ploidy levels of up to 8n have been found in sturgeon (29). GPCR duplications due to polyploidy have been proposed for the expansion of TAAR and lyso-phosphatidylserine receptors (e.g., GPR34) in the ancestor of the teleostei, the largest group of bony fishes (21, 30). A whole genome duplication likely occurred in the teleost lineage following its split from the tetrapod lineage, such that only a subset of the duplicates are retained in modern teleost genomes (31).
Ancient Records of GPCR Evolution
Whereas more than ninety-nine percent of all species that ever lived on earth are extinct, most of the genetic information about receptor repertoires and their evolutionary history must be garnered from the fossil record. Recent advances in DNA extraction and amplification from fossil remains (32) have made it possible to retrieve substantial amounts of ancient [e.g., Pleistocene (33) and Bronze age (34)] DNA sequences. Genome sequences of extant species, however, can also harbor valuable information about past functions and gene history. Pseudogenes in particular can generally be considered as genomic fossils, and they increasingly attract attention in GPCR research.
Pseudogenes are by definition non-functional genomic sequences that are homologous to a known gene (35). As a result of their non-functionality, most pseudogenes are released from selective pressure, and they therefore display an increased ratio of asynonymous to synonymous substitution rates (Ka/Ks) and accumulate frameshifting and nonsense mutations. These features are frequently used to identify pseudogenes in a genome (36). The majority of mammalian pseudogenes can be classified as duplicated pseudogenes or retrotransposed pseudogenes (also called “processed” pseudogenes). The latter are created by the reverse transcription of mRNAs followed by genomic integration. Duplicated pseudogenes arise from local duplication or unequal crossing-over; thus, they often retain, at least to some extent, the intron structures of the parental genes. Nevertheless, duplication or retrotransposition events are not always directly involved in the emergence of pseudogenes. The mutational inactivation of functional genes can be pinpointed in the genesis of specific pseudogenes, including mutational inactivation of GPCR-encoding genes (37–39).
Over time, signatures of the original sequence gradually disappear, which complicates the identification of pseudogenes (40); the exact number of pseudogenes within a complex genome can only be estimated. Rough estimates suggest that signatures of genes can be detected more than eighty million years after “pseudogenization” and loss of constraint. For example, pseudogenes for the neuropeptide Y receptor type 6 (Y6R) exist in all primates so far investigated; an inactivating deletion mutation is therefore believed to have occurred in a common ancestor of primates. Large deletions, however, can remove informative sequences in a significantly shorter period; for example, the Y6R gene disappeared in rats after the mouse/rat split fourteen to sixteen million years ago.
Pseudogenes are important because they provide molecular records of ancient genes that existed millions of years ago. Pseudogenes are particularly plentiful in the case of ORs. Whereas the human genome contains signatures of at least thirty non-olfactory rhodopsin-like GPCR pseudogenes, out of approximately 900 ORs, at least sixty-three percent appear to be pseudogenes (24). Furthermore, some apparently intact human OR genes lack motifs that are very highly conserved in their mouse orthologs, suggesting that not all human OR genes with complete open reading frames express functional OR proteins. A similar accumulation of pseudogenes has been observed in human bitter taste receptors. By contrast, in the mouse genome only about twenty percent of OR sequences are pseudogenes, giving mice over three times as many intact OR genes as humans (41). It has been speculated that the evolution of trichromatic color vision in hominoids and other Old World monkeys has relaxed the functional constraints for many taste, odorant, and pheromone receptors, thereby facilitating the development of pseudogenes (42–44).
Although the original definition of a pseudogene implied transcriptional silence, a number of reports over the years have reported pseudogene transcripts (37, 45–48). Transcripts of processed pseudogenes can contain regions with significant antisense homology, which may suggest a regulatory role for transcribed pseudogenes through an RNAi-like mechanism (49). Transcripts of several GPCR pseudogenes have been detected, but evidence for their functional relevance has yet to be substantiated.
Structural Information from Evolutionary Approaches
Currently, the only available crystal structure of a GPCR is that of rhodopsin, which provides the basis to generate structural models for all other members of the superfamily (50). Efforts to mine evolutionary trends for additional structural information have thus been crucial in modeling and mutational studies for predicting the arrangements of the seven transmembrane helices (51). To identify distinct structural determinants, such as those that participate in ligand recognition or signal transduction for a particular GPCR, sequence analysis has focused on the comparison of receptor orthologs and paralogs. The basic concept of this approach is that the structural diversity between orthologs is the result of a long evolutionary process characterized by a continuous accumulation of non-deleterious mutations. Studies based on large numbers of orthologs have analyzed the conservation and relative orientation of TMDs (21) to address the functional relevance of distinct residues in GPCRs (38, 52).
GPCR pseudogenes and their evolutionary fate can also be monitored by sequence comparisons of orthologs. An illustrative example is the chemokine receptor GPR33. Having appeared in the mammalian genome about 125–190 million years ago, GPR33 underwent parallel pseudogenization in humans, other hominoids, and some rodent species—all within just the past one million years (37). This relatively sudden occurrence across so many disparate species likely reflects a selective pressure in favor of GPR33 inactivation. A selective advantage has been established in the inactivation of the chemokine receptors CCR5 and DUFFY, which act as co-receptors for the cell entry of pathogens. CCR5 is involved in the internalization of human immunodeficiency virus-(HIV)-1, and DUFFY mediates the entry of Plasmodium vivax. Mutational inactivation of CCR5 and DUFFY leads to resistance to infection by HIV-1 (53) and P. vivax (54), respectively. Interestingly, the inactive CCR5-Δ32 allele exists in European populations at comparatively high rates (around ten percent), although the appearance of HIV-1 is too recent to explain such an allelic preponderance. Other possible selective advantages proposed for CCR5-Δ32 (55), along with non-selective scenarios (56), are still under debate.
In the case of GPR33, a cause of inactivation could be the possible interplay between receptor and a rodent-hominoid–specific pathogen; indeed, fixation of the GPR33 pseudogene is observed in rats and gerbils but not in dozens of other mammalian species (37, 38). Although the selective action of microbes upon the GPR33 pseudogene is still hypothetical, rats and gerbils are common hosts of zoonotic pathogens like hanta viruses and Yersinia pestis, and in fact both rodent species share their habitats with humans. Although evolutionary data cannot supplant functional studies of receptors, the relevance of evolutionary trends upon GPCR research is intriguing and greatly enriches our understanding of receptor repertoires.
Clinical Implications of Evolutionary Data
GPCRs play a central role in almost all physiological functions, and their involvement in pathophysiological processes is becoming clear within multiple clinical contexts. Mutations in GPCR genes are responsible for more than thirty human diseases, including cancer (57). Classical examples for GPCR-related diseases are nephrogenic diabetes insipidus (NDI) and an inherited form of obesity caused by the concomitant mutations of the vasopressin type 2 receptor (V2R) and in the melanocortin type 4 receptor (MC4R). About one third of all inactivating mutations found in GPCRs are nonsense and frame-shifting mutations, which are readily recognized upon sequence analysis. In contrast, interpreting the clinical relevance of missense mutations usually requires heterologous expression and functional characterization of the mutant receptor by ligand binding studies and second messenger assays, because missense mutations in GPCRs do not cluster in specific receptor regions. Figure 2⇓ depicts positions of missense mutations of MC4R that decrease receptor activity and are associated with inherited obesity. It is of interest to note that almost ninety percent of all functionally relevant missense mutations in MC4R (C Stäubert, T Schöneberg, H Römpler; unpublished results) and V2R (57) affect receptor sequences that have been conserved (negative selection) over 450 million years of vertebrate evolution.
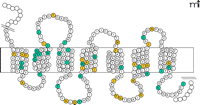
Distribution of missense mutations in the melanocortin type 4 receptor (MC4R). Like all GPCRs, the MC4R is composed of seven transmembrane domains connected by three intra- and three extracellular loops. Inactivating mutations in the MC4R are indicated in inherited forms of obesity. Amino acid substitutions at sixty positions have been found in obese humans (colored circles), but only half of missense mutations reveal a partial or complete loss of receptor function (blue circles).
Several examples demonstrate that in-depth functional characterization in combination with mining evolution as an additional source of structural information is an extremely powerful approach in the investigation of the molecular consequences of distinct disease-causing mutations in GPCRs. The likelihood that a residue is important for proper receptor function positively correlates to the degree to which the residue is conserved among vertebrates over the course of evolution. For example, most GPCRs of the rhodopsin family contain a highly conserved disulfide bond connecting extracellular loops 1 and 2. Interestingly, disease-causing missense mutations located in extracellular loops of the V2R often introduce cysteinyl residues in the expressed protein (e.g., the Y205C mutant receptor). An attractive explanation for the dysfunction is that the additional extracellular cysteine residue introduced by mutation disrupts the conserved disulfide bond or engages in other disulfide bridges. As attractive as this mechanistic explanation is, the replacement of certain wild-type residues by cysteine (e.g., the Y205C variant of V2R) can be mimicked by the introduction of residues that do not participate directly in disulfide linkages (e.g., the Y205H variant of V2R) (58), such that abolishment per se of an evolutionarily conserved and essential wild-type residue must be acknowledged.
For well-defined disease states such as NDI, the association of a phenotype with a missense mutation found in a respective GPCR can be straightforward. Obesity, in contrast, can have multiple causes; a mutation in MC4R is only one rare cause. Therefore, it is not surprising that only half of the missense variants of the MC4R found in obese humans have a partial or complete loss of function when studied in vitro (Table 1⇓). Evolutionary approaches that exploit large sets of ortholog sequence data can supplement mutational analysis of obesity. For example, the Ala175Thr substitution has been associated with obesity and positively tested for dysfunction (59–62); however, Thr at this position is naturally found in wild-type amphibian, fish, and Western European hedgehog MC4R (63) (Figure 3⇓). Similarly, the Val95Ile mutation has been identified in an obese patient and tested to be functionless, whereas Ile95 is naturally found in polar bear and armadillo MC4R, findings that prompted the re-evaluation of the human MC4R; in our hands, the Val95Ile variant of the human MC4R in manifests wild-type activity (C Stäubert, T Schöneberg, H Römpler; unpublished results). These examples nicely illustrate the power of evolutionary data to inform functional analysis and structural evaluation of disease-causing mutations in GPCRs.
aCharacterization of naturally occurring missense mutations in human MC4R

Evaluating the functional relevance of mutant MC4R by ortholog sequence comparison and functional testing. Ala175Thr within transmembrane domain 4 has been associated with obesity and positively tested for dysfunction (59–62). In amphibian, fish, and Western European hedgehog, MC4R Ala175 is naturally substituted by Thr; accordingly, the human Ala175Thr receptor protein has been examined and recently found to possess wild-type functionality (C Stäubert, H Römpler, and T Schöneberg; unpublished results).
Future Perspectives
The growing number of sequenced genomes will further support our efforts to understand phylogenetic relationships and to identify signatures of past and ongoing evolutionary selection within GPCRs. The significance and reliability of interpretations depend on both the quality and quantity of sequence data obtained from different species and populations. Extant species and populations will be the main sources for genetic information, but sequence analysis of ancient nuclear DNA will be useful for dating molecular genetic events and analyzing adaptive processes.
In GPCR research, evolution technology depends on the very robust nature of G protein–receptor crosstalk, such that signaling elements of even distantly related species (e.g., yeast and mammals) can be brought together in vitro. To enable mammalian GPCRs to activate the yeast mating pathway, for instance, only one minor change in the C terminus of the yeast G protein Gpa1p is necessary (64). Through the tailoring of the yeast G protein, activation of a mammalian GPCR can control yeast cell growth via an engineered pheromone response pathway. This coupling of receptor activity to yeast growth, moreover, allows the rapid and economical screening of randomly modified receptor libraries. For example, atropine usually acts as a blocker on M3 muscarinic receptors (M3R). However, M3Rs from libraries of millions of randomly mutated receptors expressed in yeast have been selected for growth in the presence of atropine (Figure 4⇓). Such approaches are suitable for ligand identification, mutational maturation, saturable structure-function analysis, genetic selection of constitutively active receptors, and generation of receptors with novel ligand recognition properties (65, 66).
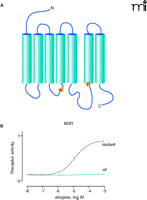
Identification of an atropine-agonized mutant M3 muscarinic acetylcholine receptor (M3R). To generate an M3R that can be activated by atropine, we applied error-prone random mutagenesis and screened approximately 1 x 107 mutant M3R clones for atropine sensitivity in a yeast expression system. In one clone we identified the mutations R179K and A489P (A) that enable the M3R to be activated by atropine (B). Both mutated positions are fully conserved within Gq-coupled muscarinic acetylcholine receptors.
Admittedly, the extraction of past evolutionary information cannot necessarily identify the selective forces that have shaped the structural evolution of GPCRs, and these past selective forces are obviously not amenable to experimental modulation and verification. However, for many biomolecules, such as RNA, enzymes, and antibodies, in vitro evolution techniques have been developed that guide structure and maturation under researcher-defined selection conditions. Over more than one billion years of GPCR evolution, the unique seven-TMD architecture has manifested discriminatory power and a remarkable degree of structural freedom to recruit almost any chemical molecule as a ligand. In keeping with the idea of a universal “receptor backbone,” artificial programming of the binding site by in vitro evolution approaches has indeed led to the generation of GPCRs with new ligand binding properties (67). Custom-designed biosensors are just one application of such evolutionary approaches.

[From left to right] Michael Hofreiter, PhD, is an independent Max Planck junior research group leader at the Max Planck Institute for Evolutionary Anthropology, Leipzig. Holger Römpler, PhD, and Angela Schulz, PhD, are postdoctoral fellows in the Institute of Biochemistry, Leipzig. Claudia Stäubert and Doreen Thor are graduate students in the laboratory of Torsten Schöneberg. Torsten Schöneberg, MD, is Professor of Molecular Biochemistry at the Institute of Biochemistry, Medical Faculty, University of Leipzig.
Acknowledgments
We would like to thank Christine Green and Adrian Briggs for many suggestions and critical reading of the manuscript. The work of the authors is supported by the Deutsche Forschungsgemeinschaft, Bundesministerium für Bildung und Forschung, the Studienstiftung des deutschen Volkes, and the Max Planck Gesellschaft.
- © American Society for Pharmacology and Experimental Theraputics 2007

