Adenylyl Cyclase Isoforms as Novel Therapeutic Targets: An Exciting Example of Excitotoxicity Neuroprotection
Recently, Wang et al. (1) demonstrated that cortical neurons from mice deficient in adenylyl cyclase type 1 (AC1) were resistant to glutamate-induced neuronal toxicity. Neuronal excitotoxicity has been linked to the activation of N-methyl-d-aspartate (NMDA) receptors (NMDARs) and appears to play a role in neuronal death associated with stroke, Huntington Disease, and other degenerative disorders (2, 3). Does this finding hold the promise of new therapies for these conditions?
The adenosine 3′,5′-monophosphate [cyclic AMP (cAMP)] signaling pathway is highly conserved among many species and is involved in a number of physiological functions in the central nervous system (4). Since the initial cloning of AC1 in 1989, eight additional membrane-bound isoforms have been cloned and char acterized (4, 5). A tenth, cytosolic (or “soluble”) AC, also has been described (6). The nine membrane-bound isoforms of AC share a similar structure that includes an intracellular N terminus, followed by two membrane-spanning domains alternating with two cytoplasmic (catalytic) domains that can be further divided into a and b regions (7). Each isoform is uniquely regulated by Gα and Gβγ subunits, divalent cations, posttranslational modification, subcellular localization, and small molecules (7–10). Group 1 ACs, represented by AC1, AC3, and AC8, are stimulated by calmodulin in a Ca2+-dependent manner. Group 2 ACs (AC2, AC4, and AC7) are characterized by their conditional stimulation by Gβγ subunits. Additionally, AC2 and AC7 are also activated by protein kinase C. Group 3 ACs (AC5 and AC6) show robust negative regulation by Gαi subunits and are also inhibited by protein kinase A and Ca2+. Group 4 is composed of AC9, which, uniquely among the ACs, is relatively insensitive to activation by forskolin.
Membrane-bound ACs are highly expressed in the central nervous system and have overlapping expression patterns (11–14). Thus, several AC isoforms are typically expressed in individual cell types, making it difficult to elucidate the function(s) of individual isoforms in either native tissues or cell lines (12, 15, 16). This problem has been addressed, however, using a variety of recombinant approaches, including overexpression, site-directed mutagenesis, and genetic deletions. Transgenic animals lacking one or multiple AC isoforms have provided significant information on the roles of AC-dependent signaling in the central nervous system (17–19).
A number of studies have been carried out with mice lacking either AC1 (AC1−/ −) or AC8 (AC8−/ −), or both isoforms. AC1 and AC8 require Ca2+ and are activated in a calmodulin-dependent fashion (20–22). They are highly expressed in neuronal tissues, including the hippocampus and several cortical regions (11, 17, 22). Early studies of long-term memory (LTM) and long-term potentiation (LTP) with animals lacking Ca2+-stimulated cyclases were driven, in part, by their high expression in the hippocampus (18). The results of these experiments implicated AC1 and AC8 in LTP and LTM (17): long lasting LTP and memory deficits were greater in animals lacking both AC1 and AC8 (double knock-out, DKO), when compared to animals deficient in only a single isoform (i.e., AC1 or AC8) (17, 18).
The development and initial characterization of the AC1−/ −, AC8−/ −, and DKO mice offered a powerful approach for identifying the molecular signaling roles for AC1 and AC8 in vivo. Further, the Ca2+-calmodulin-stimulated ACs, AC1 and AC8, are expressed in brain regions where NMDAR activation increases cAMP accumulation (23). Additionally, AC1 and AC8 may be involved in NMDA-dependent LTP, supporting the hypothesis that NMDA receptor-induced Ca2+ release could modulate the function of the Ca2+-stimulated cyclases in vivo (18). Although AC1 and AC8 were not required for the acute pain response (24), behavioral responses to inflammatory stimuli that appear to involve NMDAR pathways were markedly reduced in DKO and AC1−/ − mice (24). All of these observations are consistent with the hypothesis that NMDARs modulate the Ca2+-stimulated ACs to control physiological responses in vivo.
Thus, in the recent study by Wang et al. (1), AC1−/ − and AC8−/ − mice were compared to wild-type mice to examine the role of Ca2+-stimulated ACs in neuronal excitotoxic-ity. Glutamate treatment led to decreased cell viability and engendered markers of apop-tosis in neurons from wildtype mice, and these effects were dose-dependent and blocked by the NMDAR antagonist AP-5. The effects of glutamate on neurons from the AC8−/ − mice were essentially identical to those of wild-type mice. In contrast, cortical neurons from AC1−/ − mice were resistant to glutamate-induced neuronal toxicity at glutamate concentrations up to 250 μM. The data indicated a dramatic level (~ 90%) of neuroprotection from glutamate-induced excitotoxic-ity in the AC1−/ − neurons that could not be explained by alterations in NMDAR subunit expression or function. Further, the same pattern of neuroprotection in AC1−/ − mice was observed when cortical lesions were assessed following intracortical injection of NMDA (1).
The authors therefore proposed that AC1 is the major Ca2+ sensor for NMDAR activation and excitotoxicity (1). The selectivity for AC1, when compared to AC8, for mediating excitotoxicity is likely a result of the unique regulatory properties of each (17). For example, AC1 is sensitive to lower concentrations of Ca2+ and shows a greater synergistic response to Ca2+ and Gαs than does AC8 (17, 25). The studies from Wang et al. (1) also strongly suggest that activation of AC1, and presumably increases in cAMP accumulation, represent one component of glutamate-mediated excitotoxicity (Figure 1⇓). Stimulation of cAMP accumulation alone by activation of the serotonin subtype 7 receptor (5-HT7R), however, was not sufficient to induce excitotoxicity, which likely requires convergence with other Ca2+-modulated pathways that are activated in response to NMDAR activation (Figure 1⇓). Glutamate produced a long-lasting increase in phosphorylated cAMP response element binding protein (CREB) in neurons from AC1−/ − mice, but not from wild-type mice, which may imply that selective AC1 activation is involved in a pathway that negatively regulates CREB activity, such as Ca2+ signaling, protein phosphatases, and protein kinases (26, 27).
The results presented in Wang et al. (1) are intriguing, and the absolute specificity for the “neuroprotection” from glutamate in the AC1−/ − animals is dramatic. In contrast, an earlier study revealed no AC isoform specificity for other forms of neurodegeneration (28). Specifically, neurodegeneration induced by ethanol, MK801 (an NMDAR antagonist), or by phenobarbital was enhanced without significant AC isoform specificity in AC1−/ −, AC8−/ −, and DKO mice(28). Maas et al. also reported that the DKO animals show glutamate-induced excitotoxicity following injection of monosodium glutamate (28). That deletion of AC1 alone appears to be neuroprotective (1), whereas deletion of both AC1 and AC8 is not (28), may reflect the choice of the transgenic animals (AC1−/ − vs DKO), or the excitotoxic model (e.g., neonatal vs adult).
The genetic evidence linking AC1 to NMDA-mediated excito-toxicity does not appear to be readily explained by predicted adaptive changes involving NMDA signaling, trophic factors, or regulators of apoptosis (1, 28). Nonetheless, additional excitotoxicity studies with available animal models, including AC1-overexpressing mice, reversible or inducible knock-outs, in vivo RNAi, and the use of pharmacological agents, would provide additional supporting data. Unfortunately, few selective AC inhibitors are available. The best known are the P-site inhibitors that act by binding to the enzyme’s active site, which appears to be well-conserved among all AC iso-forms (29). Although some AC selectivity has been achieved with P-site inhibitors (30), significant concern has been raised about their specificity for other cellular processes such as DNA synthesis (31). One P-site inhibitor, SQ22536, is commercially available and cell permeable, and may have modest selectivity (fiftyfold) for AC5 (31). Based on the structure of SQ22536, a pharmacophore-based algorithm identified NKY80 as a non-adenine-containing AC5 inhibitor with increased (200-fold) selectivity for AC5 (as compared to AC2). Additional small-molecule inhibitors of AC include tyrphostin A25, calmidazolium, and catechol estrogens, all of which may share a common binding site (32–34). The binding site of these compounds is distinct from that of the P-site inhibitors and appears to differ between AC isoforms, offering an opportunity to design selective ligands (33). Nevertheless, studies examining the AC isoform specificity for these compounds using intact cells or animal models have not yet been reported.
Alternative approaches for examining function of individual AC isoforms in the central nervous system have included the use of transgenic animals overexpressing individual isoforms (35–37). In particular, future studies of mice that overexpress AC1 should provide additional insight into how AC1 participates in excitotoxicity (37). The development of selective small-molecule activators of ACs also could be very useful for dissecting the function of AC isoforms. Forskolin is a potent activator of all ACs with the exception of AC9, and forskolin derivatives with modest AC5 and AC2 selectivity and tissue-specific activity have been described (31). Nevertheless, there are very few selective AC inhibitors or activators, making it difficult to use pharmacological approaches to validate recent genetic studies. In lieu of genetic approaches, the further identification of small-molecule modulators of individual AC isoforms is an area in need of increased research, with opportunities currently being provided by NIH (38). It is anticipated that the recent studies implicating individual AC isoforms as potential drug targets will help to catalyze a closer examination of the role of ACs in cellular function and their pharmacological manipulation. Whether selective inhibitors of specific isoforms of the cyclases prove to be useful as novel treatments for neurodegenerative conditions or stroke-related injury remains to be seen, but it is an exciting possibility.
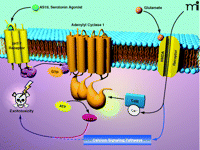
A model forN-methyl-D-aspartate receptor (NMDAR)-mediated excitotoxicity involving adenylyl cyclase type 1 (AC1). Activation of AC1 by the NMDA receptor (NMDAR), but not by the serotonin (5-hydroxytryptamine) subtype 7 receptor (5-HT7R), leads to excitotoxicity. Although the mechanism for excitotoxicity requires further study, the report by Wang et al. (1) suggests that glutamate activation of the NMDAR leads to excitotoxicity mediated through AC1, which appears to require involvement of additional Ca2+-modulated pathways (blue arrows). That is, activation of AC1 appears to be a necessary but not sufficient condition for excitotoxicity. AS19, a serotonin agonist; CaM, calmodulin; cAMP, cyclic AMP.
Acknowledgments
I would like to thank David Nichols, Benjamin Chemel, Jason M. Conley, and Julie A. Stacey for helpful comments on this manuscript. I thank David M. Allen for assistance with preparation of the figure. This work was supported by Purdue University and NIMH grant MH060397 to VJW.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Val J. Watts, PhD, is an Associate Professor of the Department of Medicinal Chemistry and Molecular Pharmacology in the College of Pharmacy at Purdue University. His research is focused on the signaling properties of G protein–coupled receptors with an emphasis on dopamine receptor systems. A long-time goal of his work has been to understand how chronic activation of inhibitory G protein–coupled receptors leads to heterologous sensitization of adenylyl cyclases in vitro and in vivo. Dr. Watts is also interested in developing small-molecule ligands as selective modulators of dopamine receptors. E-mail wattsv{at}pharmacy.purdue.edu; fax (765) 494-1414.

