Aryl Hydrocarbon Receptor Degradation-Promoting Factor (ADPF) and the Control of the Xenobiotic Response
The xenobiotic response may be defined as an adaptive function of the body to foreign chemicals, leading to the metabolism and elimination of foreign chemicals and the maintenance of chemical and functional homeostasis in the body. As humans encounter innumerable and often toxic chemicals daily from dietary, environmental, occupational, and therapeutic sources, the xenobiotic response defends the body against chemical insults. The xenobiotic response is tightly controlled in tissue- and developmental stage–dependent manners to meet the body’s demands over time. Under- or over-expression of the xenobiotic response can result in loss of the chemical homeostasis in the body, leading to disease.
The cytochromes P450 (CYPs) provide the first and critical oxygenation reaction in the metabolism of most xenochemicals (1–3). Xenobiotic-dependent induced expression of the CYPs, as well as other enzymes and proteins involved in drug metabolism and disposition (DMD), is the principal mechanism that regulates the xenobiotic response (4, 5). Increased expression of these enzymes is largely mediated through xenobiotic-activated receptors (XARs), a group of ligand-activated, structurally diverse transcription factors. In this framework, XARs sense specific chemical signals via chemical–protein interaction; activated XARs then coordinate the induction of multiple DMD genes through specific response elements located in the enhancers of the genes. Induced gene expression subsides as the inducers are metabolized by the elevated amounts of CYPs. Thus, transcription of the genes is initiated and maintained only as needed.
The aryl hydrocarbon receptor (AhR) was the first XAR identified. AhR controls the adaptive response, namely, the induction of CYP1 (1A1, 1A2, and 1B1) and several phase II enzymes, in response to the presence of polycyclic aromatic hydrocarbons [PAH, exemplified by benzo[a]pyrene (B[a]P) and 3-methyl-cholanthrene (3-MC)] and halogenated aromatic hydrocarbons [HAH, typified by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD or dioxin)] (5). Upon TCDD binding to AhR, the ligand-bound AhR translocates from the cytoplasm to the nucleus and activates the increased expression of specific genes. In addition, AhR mediates most, if not all, of the toxic responses to TCDD and HAH that include wasting (i.e., cachexia), tumor promotion, teratogenic effects, thymic involution, endocrine disruption, and skin lesions (6). Over the past half century, induction of CYP1A1, a prototypical xenobiotic response mediated by AhR, has served as a model of regulation of DMD enzymes, profoundly influencing the understanding of how small xenochemicals induce the metabolism of drugs, carcinogens, and environmental chemicals.
In the 1950s, the induction of CYP1A1 was discovered in an attempt to understand the mechanism by which low doses of 3-MC delay or totally block the carcinogenic and hepatotoxic effects of 3′-methyl-4-dimethylaminoazobenzene (3′-Me-DAB), a potent hepatocarcinogen, in rats (7, 8). The effects of 3-MC and other PAH on the hepatic N-demethylation and azo-link reduction of aminoazo dyes, metabolic pathways resulting in noncarcinogenic products, were examined. PAH that inhibit aminoazo dye–induced liver cancer were recognized as potent inducers of azo dye N-demethylase, whereas PAH that did not affect azo dye–induced carcinogenesis had little or no effect on azo dye metabolism, providing the first evidence of CYP induction and its large impact on chemical carcinogenesis and toxicity (8). Later, 3-hydroxylation of B[a]P—a reaction catalyzed by aryl hydrocarbon hydroxylase [(AHH), an enzymatic activity of CYP1A1 (also termed cytochrome P448)]—was used as a standard assay of PAH-inducible CYP activity; the induction of AHH expression and activity subsequently became integrated in the mechanistic model of CYP regulation, partly because of the essential role of AHH in activating B[a]P to the ultimate carcinogen trans-7,8-diol 9,10-epoxide (9).
Induced AHH expression in inbred mouse strains provided genetic evidence that led to the identification of the AhR. Mouse strains such as C57BL/6 (B6) are sensitive to AHH induction by 3-MC, whereas strains such as DBA/2 (D2) are resistant to the induction. The sensitive phenotype segregates as a single autosomal dominant trait, defining a genetic locus designated as the Ah locus (aromatic hydrocarbon responsiveness); Ahb represents the Ah responsive genotype (i.e., the B6 allele) whereas Ahd represents the Ah non-responsive genotype (i.e., the D2 allele) (10, 11). Comparative studies of the potency and specificity of AHH inducers (B[a]P, 3-MC, TCDD, and congeners) for AHH induction in Ahb and Ahd strains—and later, the demonstration of reversible, saturable, and high-affinity binding of radiolabeled TCDD to a soluble cytoplasmic protein in the Ahb mouse liver—led to the proposal that the Ah locus encodes the Ah receptor (AhR) that binds the inducers and initiates AHH induction (12–14).
Induction of AHH expression by B[a]P in hepa1 cells, a mouse hepatoma cell-line strongly responsive to AHH inducers, is detrimental to the cells because of increased formation of trans-7,8-diol 9,10-epoxide and B[a]P-related DNA adducts. This AHH-dependent suicidal response was exploited genetically to dissect the major components of the signal transduction involved in inducing AHH expression by selecting B[a]P-resistant variant cells in culture (15). At the same time, fluorescence-activated cell sorting (FACS) was utilized to isolate variant cells with either a markedly elevated or markedly decreased ability to metabolize B[a]P (16). Variants with similar phenotypes of AHH induction were obtained with both approaches (17, 18). Notably, some cytoplasmic extracts of cell variants exhibited reduced binding of AhR to TCDD, others had aberrant nuclear translocation of AhR, and still another cell group had normal TCDD binding and comparable nuclear translocation of AhR but no AHH activity. Complementation analyses separated the variants into at least three groups, which were later defined as the genes encoding CYP1A1, AhR, and the Aryl hydrocarbon receptor nuclear translocator (Arnt), respectively. Parallel to the genetic analysis of the hepatoma variants, studies of the promoter region of mouse Cyp1a1 led to the identification of multiple DNA sequences that share a core recognition element of 5′ T-GCGTG 3′ (where the hyphen indicates any nucleotide base may be present) (19). These sequences can promote AhR agonist–mediated expression of a reporter gene and form DNA-protein complexes with AhR and Arnt as shown by gel retardation assay in vitro. The core sequence was defined as a “dioxin responsive element” (DRE), representing the binding sites of AhR and Arnt in the enhancer of Cyp1a1.
Subsequently, efforts were focused on cloning the components of induction. AhR was purified from B6 mouse liver to apparent homogeneity by covalently labeling AhR with a radiolabeled photoaffinity ligand, followed by chromatography (20). The N-terminal sequence derived from the purified AhR (Ahb−1 allele) was utilized to clone the cDNA of mouse AhR, which was identified as a basic helix-loop-helix transcription factor (21). Arnt, which is the DNA-binding partner of AhR, was cloned by complementation of Arnt-deficient cell variants with human genomic DNA, followed by reverse selection for AHH induction (22). Arnt shares a modular domain structure similar to that of AhR. The cytoplasmic AhR exists in a complex with at least two molecules of hsp90 (23) and the AhR-interacting protein (AIP) (24), a multifunctional and tetratricopeptide repeat (TPR) motif–containing chaperon molecule (Figure 1⇓). This complex maintains the receptivity of AhR for ligand. Thus, a prototype transcriptional circuit of a cytoplasmic XAR for CYP induction is constructed: XAR is silenced by sequestration in the cytoplasm; binding with an agonist activates XAR; activated XAR translocates into the nucleus and heterodimerizes with a partner protein; and the XAR dimer binds to specific DNA sequences located in the enhancers of a battery of DMD genes.
A critical question concerning Cyp1a1 induction arises from the early characterization of AHH induction: Is AhR-mediated induction controlled at a secondary level beyond the AhR-ligand circuit in cells? The answer is clearly, “Yes.” In cultured cells, pre- or co-treatment with the protein synthesis inhibitor cycloheximide (CHX) in the presence of an AhR agonist, followed by CHX wash-out and continued culture (to allow protein synthesis), increases AHH induction six- to tenfold relative to that observed with treatment with the AhR agonist alone (25). Increased AHH induction in the presence of CHX and AhR agonists results from increased transcriptional rate and, hence, increased formation of Cyp1a1 mRNA and protein but not from increased stability of the mRNA or protein (26, 27). This phenomenon of increased Cyp1a1 mRNA induction is termed “superinduction.” Superinduction requires substantial inhibition of cellular protein synthesis (>90%), suggesting that the synthesis of a repressor protein, which is either short-lived (labile) or is induced during Cyp1a1 induction, or both, is inhibited (28). Superinduction requires functional AhR, as superinduction is observable but largely reduced in AhR-deficient variants and is absent in Arnt-deficient variants that fail to accumulate AhR in the nucleus in response to an agonist (26, 29). CHX does induce Cyp1a1 in the absence of an exogenous AhR agonist (albeit to a much smaller extent than that observed in superinduction); however, CHX-mediated induction depends upon functional AhR and Arnt. Two conclusions can be drawn from these observations: 1) induction of Cyp1a1 via AhR is a primary response (i.e., it does not require new protein synthesis), and 2) induction is repressed by a CHX-sensitive protein such that transcription of Cyp1a1 peaks an order of magnitude lower relative to normal conditions. The repression of induction suggests a secondary mechanism of transcriptional Cyp1a1 regulation in which a CHX-sensitive repressor modulates the agonist–AhR complex to control Cyp1a1 induction. These mechanisms ensure that increased gene expression is adequate and flexible to the physiological need of cells but can be terminated promptly.
The nature of the CHX-sensitive repressor has been elusive. Gel retardation analyses of nuclear extracts and methylation protection studies in intact cells revealed no evidence for CHX-sensitive protein–DNA interactions that might mediate the superinduction response (28). Superinduction also occurs in other AhR agonist–dependent gene expression, such as the TCDD-inducible poly(ADP-ribose) polymerase (TiPARP) (29). Together, these observations negate the possibility of the repressor as a DNA-binding protein, as a component of the AhR–DRE complex, or as a modulator of chromatin configuration that may affect AhR accessibility to DNA. On the other hand, they are consistent with a mechanism in which the CHX-sensitive repressor modulates AhR or a component of the AhR pathway via protein-protein interaction to inhibit AhR function.
The availability of antibodies specific for AhR has made it possible to directly characterize AhR as a stable protein in unstimulated cells (t1/2 = 28 hrs), consistent with the observation that AhR-mediated Cyp1a1 mRNA induction does not require new protein synthesis (30). Treatment with TCDD, however, induces rapid turnover of AhR protein (t1/2 = 3 hrs). The agonist-induced degradation of AhR is mediated through the ubiquitin-26S proteasome pathway, as evidenced by the following observations: 1) degradation is inhibited by proteasome inhibitors MG132 and lactacystin; 2) TCDD induces ubiquitination of AhR that is further enhanced by MG132 treatment; 3) degradation is E1-dependent; and 4) MG132 increases the amount of AhR protein and AhR–DNA complex formation in the nucleus, suggesting agonist-activated nuclear AhR as the target of degradation (30). Surprisingly, a connection between agonist-induced AhR turnover and superinduction of Cyp1a1 was realized when CHX was shown to inhibit AhR degradation under conditions that promote superinduction (31). Thus, CHX (like MG132) blocks TCDD-induced shortening of the half-life of AhR; inhibition of AhR degradation correlates with superinduction in both time- and concentration-dependence of CHX treatment; and CHX increases the amounts of AhR and the AhR–DRE complex in the nucleus. Together, these findings underpin the hypothesis that the CHX-sensitive repressor mediates or controls the ubiquitination–proteasomal degradation of nuclear AhR; inhibition of the synthesis of the repressor blocks the degradation (and thereby increases the amount) of activated AhR in the nucleus, resulting in increased transcription of target genes. The CHX-sensitive factor has been designated AhR degradation–promoting factor (ADPF) (31) (Figure 1⇓).
Protein degradation through the ubiquitin-26S proteasome pathway consists of polyubiquitination of a target protein and proteasomal degradation of the ubiquitinated protein. The specificity of protein ubiquitination is mostly controlled by target-specific E3 ubiquitin ligase. As an effective way of terminating the function of a specific protein, ubiquitin-proteasomal degradation has been broadly implicated in cellular functions including cell division and growth, differentiation, transcriptional gene regulation, removal of damaged proteins, and senescence [see (32)]. Proteasomal degradation regulates transcription factors at both the activation and termination phases of transcription. Degradation serves as a mechanism of silencing labile transcription factors, such as c-Myc, p53, AP-1 proteins, and Nrf2, in unstimulated cells. Signal-induced inhibition of the degradation, which often involves inhibition of a pathway-specific E3, leads to the stabilization and activation of the transcription factor. Alternatively, signal-induced degradation of inhibitory subunits of a quiescent transcription factor complex can reslut in the activation of transcription, such as in the case of LPS activation of nuclear factor–κB (NF-κB) by inducing ubiquitin-proteasomal degradation of the inhibitor of NF-κB–α (IκBα).
The prominent role of proteasomal degradation in the termination of transcription has only been recognized in recent years (30, 33). In one example, DNA damage induces and activates p53 to induce cell cycle arrest and apoptosis; at the same time, Mdm2, an E3 for p53, is induced by p53 to catalyze the ubiquitination of p53 leading to p53 degradation and to the termination of p53-mediated signals (34). In this manner, transcription and ubiquitin-proteasomal degradation are coupled to regulate the amount of p53 protein present and, consequently, the extent and duration of the p53 response to DNA damage. The functional importance of Mdm2-mediated regulation of p53 is indicated by the finding that deletion of mdm2 is lethal during early embryogenesis in mice carrying a functional p53 gene. In another instance, PDLIM2, a PDZ and LIM domain–containing E3, promotes the ubiquitination and targeting of the 65-kDa subunit (p65) of (NF-κB) to “promyelocytic leukemia” (PML) nuclear bodies where proteasome-directed degradation of p65 occurs. PDLIM2 deficiency results in larger amounts of nucleus-localized p65, defective p65 ubiquitination, and augmented production of proinflammatory cytokines in response to innate stimuli (35).
By analogy with Mdm2 and PDLIM2, we propose that ADPF functions as an E3 or E3 regulator that controls the ubiquitination of nuclear AhR (Figure 1⇓). This hypothesis is supported by the observation that CHX blocks TCDD-induced ubiquitination of AhR (31). The sensitivity of ADPF to CHX can be explained by two alternative but not mutually exclusive mechanisms. ADPF may be an AhR target protein that is inducible by TCDD. Alternatively, ADPF could be labile, requiring constant protein synthesis for function. The observation that pretreatment with TCDD (>10 hrs) before CHX treatment also superinduces Cyp1a1 supports the conclusion that ADPF is a labile protein (28). Direct proof of the hypotheses will require the cloning of ADPF. Recently, AhR was reported to function as an adaptor that brings estrogen receptor (ER) to a cullin 4B (Cul4B)-based ubiquitin ligase complex and thereby targets sex steroid receptors for degradation. In this model, AhR serves as an atypical component of the E3 of ER (36). These findings provide a mechanistic explanation to certain endocrine disruptor activities of TCDD and AhR by way of promoting ER degradation. Whether this unconventional Cul4-E3 plays a role in agonist-induced AhR turnover or is related to the function of ADPF remains to be determined.
Aberrant function of ubiquitin-26S proteasome pathways is often associated with diseases in humans ranging from cancer, chronic inflammatory disease, and muscle wasting, to neurodegenerative disorders and aging. Accordingly, ubiquitin-proteasomal regulation of protein turnover has been utilized as therapeutic targets of human disorders. Because of the high specificity of E3 for target proteins, the advantage and feasibility of targeting particular E3s for specific pathway(s) in drug development are being increasingly recognized, such as in the case of chemoprevention by inhibiting the Cul3/Keap1-dependent E3 that targets Nrf2, and in the treatment of cachexia in cancer patients by inhibiting MuRF1 and MAFbx, two E3s activated during muscle wasting.
The demonstration of the interconnection between TCDD-induced AhR turnover and superinduction of Cyp1a1 underscores the importance of controlling the amount of AhR protein in the nucleus. Inhibition of this control mechanism, such as by inhibiting ADPF, would likely elevate AhR activity and exacerbate the carcinogenic and toxic effects of PAH and HAH. It is also conceivable that ADPF or the proteasomal degradation of nuclear AhR could serve as a target for developing therapeutic and preventive drugs for cancer and other diseases related to AhR and its ligands. One advantage of such drugs would be that they are pathway-specific for the AhR function rather than targeting individual CYPs. Given that drug metabolism is largely regulated transcriptionally through XARs, regulation of XARs by proteasomal degradation in the nucleus could serve as a general means of controlling the xenobiotic response, analogous to agonist-induced AhR turnover. Whether ADPF plays a role in the ubiquitin-proteasomal degradation of XARs other than AhR may be tested in future. If ADPF does regulate multiple XARs, this might allow for the development of one drug that modulates the activities of several XARs at one time. Becuase XARs generally regulate multiple DMD enzymes, ADPF inhibitors or boosters could be effective and versatile modulators of more DMD activities than can current CYP inhibitors. Whether this multiplicity of targets would be therapeutically warranted, however, awaits further investigation.
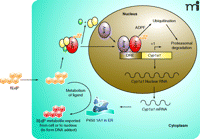
Induction of Cyp1a1 and ADPF-controlled degradation of nuclear AhR. In the absence of an agonist, the quiescent AhR is in a complex with two molecules of hsp90 (yellow ovals) and the AIP protein (green oval) in the cytoplasm; B[a]P-binding activates AhR, which translocates into the nucleus and dimerizes with Arnt; the AhR-Arnt dimer binds to a dioxin responsive element (DRE) and initiates the transcription of Cyp1a1. B[a]P-activated AhR is degraded through the ubiquitin-26S proteasome pathway that is controlled by ADPF (as a probable E3) in the nucleus. Cyclohexmide (CHX) blocks the synthesis of ADPF and consequently the B[a]P-induced degradation of nuclear AhR, resulting in superinduction. ADPF, AhR degradation promoting factor; AhR, Aryl hydrocarbon receptor; AIP, AhR-interacting protein; B[a]P, benzo[a]pyrene; Arnt, Aryl hydrocarbon receptor nuclear translocator; ER, endoplasmic reticulum.
Acknowledgments
The interpretations, discussions, and conclusions in this report are those of the author and do not necessarily represent the views of the National Institute for Occupational Safety and Health.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Qiang Ma, MD, PhD, is a Lead Toxicologist and Team Leader at NIOSH, CDC, and also an adjunct professor in the Department of Biochemistry and Molecular Pharmacology, West Virginia University School of Medicine. He has focused on how small chemicals regulate xenobiotic response transcriptionally via xenobiotic-activated receptors (XARs) and the roles of XARs in chemical toxicity, drug-drug interaction, and disease pathogenesis. He was the recipient of the 2007 ASPET Division for Drug Metabolism Early Career Achievement Award and the 2007 NIOSH Alice Hamilton Award in Biological Science Category. E-mail qam1{at}cdc.gov; fax 304-285-5708.

