Novel Insights into the Beneficial and Detrimental Actions of Cdk5
Cyclin-dependent kinase 5 (Cdk5) is a serine–threonine kinase with remarkably diverse functions. Although Cdk5 does not regulate the cell cycle as the name suggests, it has critical functions in post-mitotic neurons (1–3). In the adult brain, Cdk5 can be beneficial by contributing to memory formation or it can be detrimental by causing neurodegeneration, and it is of great interest to understand this dichotomy. Cdk5 is activated by the neuron-specific co-activators p35 and p39. Interestingly, under pathological conditions, both of these co-activators can be proteolytically cleaved by the Ca2+-sensitive protease calpain to the smaller, more stable proteins, p25 and p29, respectively, that over-activate Cdk5.
About five years ago, it became apparent that Cdk5 is involved in memory formation (2). Inhibitors of Cdk5 were shown to impair hippocampus-dependent contextual fear conditioning (4) (Box 1). Furthermore, low levels of p25 expression that increase Cdk5 activity concomitantly enhance hippocampus-dependent spatial memory formation (5–7). Transiently induced expression of p25 in transgenic mice confirms the enhancement of hippocampal memory formation (8). Importantly, low amounts of p25 expression do not always enhance memory formation; instead they can impair memory formation when training is very complex (9). This finding indicates that p25-mediated over-activation of Cdk5 acts as cognitive enhancer rather than as a general performance enhancer. Additionally, global p35 knock-out mice with reduced Cdk5 activity are impaired in spatial memory formation, although this result might be caused by the developmental abnormalities rather than effects on synaptic plasticity (10). Taken together, there is good evidence that Cdk5 plays a critical role in memory formation, but it remains largely unknown which mechanisms are regulated by Cdk5. It is conceivable that Cdk5 contributes to synaptic plasticity because the kinase has various synaptic functions in vitro (2, 3) and in hippocampal slices it is required for long-term potentiation (LTP) (11), the prime model of memory formation and storage. Cdk5 might also contribute to the formation of dendritic spines, which are believed to contribute to memory formation (8); however, more investigations are needed to identify the role of Cdk5 in cognition. The recent study by Hawasli et al. (12) is a significant step toward this direction.
The Role of the Hippocampus in Learning and Memory
In the 1950s, it became known that the hippocampus plays a critical role in human memory. At that time, the hippocampus was removed from patient H.M. to treat severe epilepsy. The treatment prevented the epilepsy, but it resulted in severe deficits in memory formation, affecting specifically declarative memory––memory one is aware of. Thus, the hippocampus plays a key role in memory formation and it is interesting to note that hippocampus-dependent memory declines in the early stages of Alzheimer Disease. Systematic lesion studies in animals have demonstrated that the hippocampus is essential for several types of memory, including spatial memory. In contextual fear conditioning, animals are tested to determine whether they can make an association between an environment (context) and an aversive stimulus; this association requires the hippocampus.
Hawasli et al. (12) used conditional knock-down mice to investigate the role of Cdk5 in synaptic plasticity and behavioral learning and memory. This approach complements the previous Cdk5 inhibitor and p25 expression studies, the latter of which showed that p25 over-activates Cdk5. Conditional knock-down of Cdk5 enhances contextual fear conditioning and improves learning of a new spatial location after a spatial memory has been formed, which is called spatial reversal learning. This finding is in contrast to what was expected from previous inhibitor and p25 expression studies (4–6, 8). As explained below, Cdk5 has not only a role as a kinase but also as a scaffold protein. It is interesting to note that, in contrast with the studies on memory formation, the results of the role of Cdk5 in memory extinction converge: The knockdown of Cdk5 activity enhances extinction of contextual fear memory, which has also been observed with the administration of Cdk5 inhibitors and which is in agreement with p25 expression studies (12, 13).
To address the cellular and molecular bases of improved memory formation in the Cdk5 knock-down mutants, Hawasli et al. performed electrophysiological recordings at CA1 synapses in hippocampal slices. They observed enhanced N-methyl-d-aspartate (NMDA) receptor currents were enhanced and a reduced threshold of LTP induction was reduced (12). These changes provide an intuitive explanation for enhanced hippocampus-dependent memory formation, because less stimulation can induce LTP in the Cdk5 mutants. Consistent with this finding, mice harboring other genetic deletions resulting in reduced threshold of LTP induction also have improved hippocampal memory formation [see (14)]. Further, Hawasli et al. established that the increased expression of the NMDA receptor subunit NR2B accounts for the LTP change (12). Interestingly, the increase in NR2B protein expression is not accompanied with augmented mRNA expression. Subsequently, p35, Cdk5, NR2B, and calpain were shown to form a complex and that the calpain degrades NR2B (Figure 1⇓). Thus, the knock-down of Cdk5 protein expression prevents degradation of NR2B protein without affecting NR2B mRNA expression. Importantly, the degradation of NR2B does not depend on the kinase activity of Cdk5 but rather is a result of a protein-protein interaction with Cdk5 and calpain. Taken together, Hawasli et al. have uncovered a mechanism, the calpain-directed degradation of NR2B, which can explain how Cdk5 contributes to memory formation. For this mechanism to be of physiological relevance it is important that training in a behavioural memory task regulates the expression of Cdk5 protein as shown after contextual fear conditioning (4). Furthermore, it should be noted that Cdk5 can contribute to memory formation because of its kinase function and that alteration of Cdk5 activity (rather than Cdk5 protein amount) also matters, as the Cdk5 inhibitor studies and p25 expression studies have demonstrated. It will be important to establish how altered Cdk5 activity impacts on memory formation and whether the kinase activity is more important than the regulation of NR2B expression by Cdk5.
p25-mediated overactivation of Cdk5 is not always beneficial for the brain because under certain conditions, excess amounts of p25 cause neurodegeneration (8, 15). Neurodegeneration triggered by Cdk5 over-activation has been suggested to occur in both Alzheimer and Parkinson Diseases (16, 17). Currently, not much is known as to how overactivation of Cdk5 causes neurodegeneration. Hyperphosphorylation of the cytoskeletal protein tau has been widely considered a key step, and Cdk5 hyperphosphorylates tau at sites that are hyperphosphorylated in Alzheimer Disease. A recent study, however, suggested that glycogen synthase kinase 3 (GSK3), rather than Cdk5, is the key player for tau hyperphosphorylation (7). Moreover, an investigation of a model of Parkinson Disease proposed that Cdk5 over-activation inhibits the transcription factor myocyte enhancer factor 2 (MEF2), leading to loss of dopaminergic neurons (18). Thus, it remains unclear how important the nuclear role of Cdk5 is in neurodegeneration.
Recently, Qu et al. (19) uncovered important insights as to how Cdk5 overactivation in the cytoplasma can cause neurodegeneration. Qu et al. used the mitochondrial toxin MPTP to model neurodegeneration related to Parkinson Disease. In earlier studies, these authors showed that MPTP activates calpain resulting in the formation of p25 and overactivation of Cdk5 (17, 18). Qu et al. screened for a cytoplasmic target of Cdk5 activation and found that the antioxidative enzyme peroxiredoxin-2 (Prx2) associates with Cdk5 and p35. This association is important insofar as follow-up studies revealed that both p25-Cdk5 and p35-Cdk5 can phosphorylate Prx2. Qu et al. used in vitro and in vivo models to show that Cdk5 phosphorylates Prx2 at threonine-89 (Thr89) and that this phosphorylation inhibits Prx2 activity. Inhibition of Prx2 activity leads to increased concentrations of reactive oxygen species (ROS) and causes neuronal death. In contrast to the inhibition of Prx2, overexpression of Prx2 appears neuroprotective. Finally, these authors showed that Prx2 phosphorylation at Thr89 is increased in post-mortem tissue from patients with Parkinson Disease. Taken together, these studies uncovered a critical role for the regulation of Prx2 activity by p25-Cdk5 and p35-Cdk5 in neurodegeneration related to Parkinson Disease. It remains to be tested whether dysregulation of Prx2 activity can also account for other types of neurodegeneration that are p25-Cdk5–dependent.
Cdk5 is a remarkable kinase because it has multiple functions, some of which are beneficial and some of which are detrimental for the adult brain. In fact, the hypothesis was proposed that formation of p25 (which overactivates Cdk5) is a compensation for early learning deficits in Alzheimer Disease and that continued p25 compensation leads to neurodegeneration (5, 6, 8, 15). Consequently, it would not be appropriate to treat neurodegeneration by simply blocking Cdk5 activity [see (7)]. Instead, it is essential to identify relevant Cdk5-dependent signaling pathways to find specific targets for molecular interventions. Finding of such targets and interventions holds promise for the enhancement of cognition and for neuroprotection. The recent studies by Hawasli et al. and Qu et al. are first milestones towards this direction.
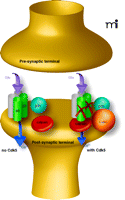
Cdk5 protein regulates calpain-directed degradation of the NMDA receptor subunit NR2B. p35-Cdk5 binds calpain, which then degrades NR2B. The degradation of NR2B does not occur in the absence of Cdk5 protein. Smaller arrow (found in conditions where Cdk5 is present) indicates diminished signaling through NMDA receptors; absence of NR2B subunits precludes signaling by these receptors. Thus, the overall signal transduced by the total number of ligand-bound NMDA receptors is reduced.”
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Karl Peter Giese, PhD, is Professor of Neurobiology of Mental Health at the Centre for the Cellular Basis of Behaviour at the Institute of Psychiatry at King’s College London, UK. He is also an Associative Member of the MRC Centre for Neurodegeneration Research at King’s College London. His research concentrates on the molecular analysis of function and dysfunction in memory processes in mouse models. He is studying the roles of Cdk5 and Ca2+/calmodulin-dependent kinase II, and he is interested in the molecular basis of long-term memory formation and storage. E-mail Peter.Giese{at}iop.kcl.ac.uk; fax +44 2078485308.

