Phase 0 Clinical Trials in Cancer Drug Development: From FDA Guidance to Clinical Practice
- Robert Kinders1,
- Ralph E. Parchment1,
- Jay Ji1,
- Shivaani Kummar2,
- Anthony J. Murgo3,
- Martin Gutierrez2,
- Jerry Collins3,
- Larry Rubinstein3,
- Oxana Pickeral3,
- Seth M. Steinberg2,
- Sherry Yang3,
- Melinda Hollingshead3,
- Alice Chen3,
- Lee Helman2,
- Robert Wiltrout2,
- Mel Simpson1,
- Joseph E. Tomaszewski3 and
- James H. Doroshow2,3
- 1Laboratory of Human Toxicology and Pharmacology, Applied/Developmental Research Support Directorate, SAIC-Frederick Inc., NCI-Frederick, Frederick, Maryland, 21702;
- 2Center for Cancer Research, National Cancer Institute, Bethesda, Maryland, 20892;
- 3Division of Cancer Treatment and Diagnosis, National Cancer Institute, Bethesda, Maryland, 20892
Abstract
The Food and Drug Administration (FDA) recently introduced the Exploratory Investigational New Drug Guidance to expedite the clinical evaluation of new therapeutic and imaging agents. Early clinical studies performed under the auspices of this guidance, so-called “Phase 0” trials, have been initiated at the National Cancer Institute to integrate qualified pharmacodynamic biomarker assays into first-in-human cancer clinical trials of molecularly targeted agents. The goal of this integration is to perform molecular proof-of-concept investigations at the earliest stage of cancer drug development. Phase 0 trials do not offer any possibility of patient benefit; instead, intensive, real-time pharmacodynamic and pharmacokinetic analyses of patient tumor samples and/or surrogate tissues are performed to inform subsequent trials. Phase 0 studies do not replace formal Phase I drug safety testing and require a substantial investment of resources in assay development early on; however, they offer the promise of more rational selection of agents for further, large-scale development as well as the molecular identification of potential therapeutic failures early in the development process.

Introduction
In recent years, much attention has been drawn to the numerous challenges facing developers of new anticancer drugs. The expected boom in the availability of novel molecules has not resulted from the completion of the human genome project. Increases in both the time required for drug development and the complexity of clinical trials utilizing advanced technologies, as well as the low predictability of toxicity and efficacy based on traditional animal models, have all led to higher costs as well as greater risk aversion by an increasingly consolidated pharmaceutical industry (1, 2).
Lack of efficacy has emerged as the leading cause of attrition in clinical trials of novel therapeutics, with less than ten percent of Investigational New Drug (IND) applications for novel molecules moving beyond the earliest stage of development (1). Innovative oncologic drug development has also been affected by the lack of novel biomarkers that would allow regulatory approvals to advance more rapidly based on validated biomolecular signals that correlate specifically with early clinical endpoints (3).
This situation emphasizes the urgent requirement to streamline and focus the development of novel cancer therapeutics at the national level. Drug development timelines need to be shortened in concert with enhanced molecular drug discovery efforts. There is also an urgent need to establish processes to assess anticancer drug action (i.e., safety, efficacy, and mechanism of action in vivo) much earlier in the drug development cycle, thus avoiding expensive failures during late-phase studies; and to provide a rigorous, and more effective, scientific basis for the wide variety of potential indications for new oncologic drugs. Ultimately, there is a critical need to more effectively apply currently-available molecular tools to the oncologic drug development process to accelerate productivity and enhance innovation, speeding the delivery of novel anti-cancer agents to the bedside.
Among the current challenges in oncology drug development is the inability to predict which new drugs will have clinical efficacy prior to human testing. Traditional first-in-human (Phase I) studies are conducted to evaluate drug safety and tolerability and establish the maximum tolerated dose (MTD) for subsequent Phase II safety monitoring and human pharmacokinetics studies. The initial Phase I dose is based on the severely toxic dose (STD) or MTD in the most sensitive animal species identified during extensive preclinical toxicology testing (4). Drug “success” warranting submission of a New Drug Application (NDA) to the FDA is not established unless efficacy can be demonstrated in Phase III trials. Reaching this milestone requires the participation of large numbers of patients and the expenditure of considerable resources. Since current approval rates for new oncology drugs are estimated to be no more than 5%, a mechanism that identifies efficacy earlier in the process would eliminate “failures” and focus effort only on the most promising new agents (1, 5, 6).
In 2006, the US Food and Drug Administration (FDA) addressed some of these early drug development issues with the issuance of the exploratory Investigational New Drug (IND) Guidance (7). Developed in conjunction with the pharmaceutical industry and the National Cancer Institute (NCI), the exploratory IND allows pilot clinical studies of new drugs and imaging agents administered in limited doses to a small number of patients without therapeutic or diagnostic intent. The preclinical pharmacology and toxicology testing required for an exploratory IND is less extensive than for a traditional IND because the FDA considers that the doses and dosing schedules to be used confer a lower potential risk of toxicity. There are differences in the preclinical and clinical study pathways for traditional and exploratory IND applications (Figure 1⇓). The reduced preclinical study package required for new agents evaluated under the exploratory IND allows clinical evaluation much sooner than would be expected under a traditional IND (Figure 2⇓).
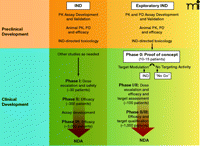
Key components of cancer drug development from preclinical evaluation through NDA submission following traditional (Phase I) or exploratory IND (Phase 0) clinical pathways. The objective of preclinical studies performed to support a traditional IND is to estimate a starting dose for Phase I trials which is both safe and allows for the possibility of clinical benefit. Subsequent Phase II and III trials continue to monitor drug safety and bioavailability as well as evidence of efficacy. Large numbers of patient volunteers are needed at each clinical stage, and the drug can fail for unacceptable toxicity, poor pharmacokinetics, or lack of efficacy at any point during this pathway. The objective of Phase 0 trials is to anticipate clinical failure and potential efficacy. Study endpoints focus on target modulation, necessitating development and validation of plasma drug (pharmacokinetic) and preclinical biomarker (pharmacodynamic) assays during preclinical evaluation. If the drug shows promise, the experimental IND is closed and further development proceeds under a traditional IND to explore drug safety. Phase I/II trials (target assessment studies) of molecularly-targeted agents can then be performed with a qualified pharmacodynamic assay that could enhance the basis on which future development decisions for the agent are made. Progression to Phase II/III randomized trials would then occur with a stronger scientific base. PK, pharmacokinetics; PD, pharmacodynamics; NDA, new drug application; IND, investigational new drug.
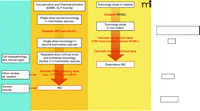
Schema outlining the preclinical testing strategy for traditional and exploratory INDs. Under the traditional IND, for other therapeutic areas, safety pharmacology studies are conducted to address the systemic effects of a new agent on vital cardiovascular, CNS, and respiratory systems; these studies can be conducted as stand-alone trials or as a component of later animal studies. The FDA may not require the conduct of safety pharmacology studies under an exploratory IND if the study involves a single microgram-quantity dose for imaging or PK analysis. In oncology, safety pharmacology studies are not required prior to filing of an IND, exploratory or traditional. Other studies generally conducted under a traditional IND include pharmacokinetic and pharmacology studies to evaluate different dosing schedules or routes of administration, such as tests of animal absorption, distribution, metabolism, and excretion (ADME), analytical assays, and drug formulation testing. Both INDs require single-dose (acute) toxicology studies in two mammalian species to determine toxic and safe doses. For the exploratory IND, these studies may have a single dose or dose escalation design; subsequent repeat-dose toxicology studies in oncology reflect the proposed clinical schedule. For the traditional IND, two mammalian species are tested for cumulative toxicities arising from repeat-dosing that reflects the clinical route and schedule, including full histopathology and clinical sign evaluation; other preclinical studies to assess drug or vehicle activity and safety and support the IND can be conducted as needed. Genetic toxicity testing of oncology agents is not necessary until before Phase II testing. Full genetic toxicity evaluation includes in vitro bacterial reverse mutagenicity tests, in vitro mammalian chromosome damage tests, and in vivo chromosomal damage tests (i.e., in vivo rodent micronucleus assay). MTD, maximum tolerated dose; NOAEL, no observed adverse effect level; 1/10 STD10, one-tenth of that dose which causes severe toxicity or death (STD) in 10% of animals; GLP, good laboratory practices.
Primary study endpoints of trials conducted under an exploratory IND can include: 1) evaluations of analogs for lead selection; 2) modulation of a molecular target (i.e., change in gene or protein expression) in a tumor in vivo; 3) whole-body imaging for tissue distribution and target binding affinity; and 4) agent pharmacokinetics (PK). To accomplish this, the study design must integrate measures to quantify drug effect, for example a pharmacodynamic (PD) assay of whether the agent inhibits a specific enzyme, to allow rational decisions about further drug development (8, 9). The exploratory IND is intended to speed the process of drug development by providing opportunities to refine the lead optimization process in vivo; however, promising drugs will still need to be evaluated for toxicity and efficacy under a traditional IND (7).
Having participated in the development of the exploratory IND, investigators at the NCI recognized the necessity of proof-of-concept testing for molecularly targeted drugs. The NCI began conducting Phase 0 trials under the exploratory IND guidance early in 2006 (6, 10). It is anticipated that investing in the Phase 0 process will help the NCI perform its mission of enhancing the entry of promising early-stage drug candidates into its therapeutic pipeline. Specifically, Phase 0 trials may help to eliminate drugs that are likely to fail later-stage efficacy testing well before moving into trials that require large numbers of patients to establish drug tolerability and safety. The goals of this effort are to identify promising agents earlier, develop and establish PD assays in human samples prior to instituting larger trials, and possibly shorten the drug development timeline. Accomplishing these goals should increase the success rate of new agents entering clinical development and bring active oncology drugs to market faster. One additional advantage of this approach is the increased probability that a patient participating in a late-phase trial will experience clinical benefit. Preclinical effort will be directed at identifying predictors of response to molecularly-targeted agents so that patients can be screened prior to treatment. Standard operating procedures (SOPs) developed for every aspect of sample collection, processing, and testing will also ensure that all patient samples collected for analysis give reproducible and clinically useful results.
Initiating a Phase O Trial: Differences in Dosing
Historical guidelines recommend that the starting dose for Phase I clinical trials in oncology be one-tenth of that dose which causes severe toxicity or death (STD) in 10% of animals—generally 2) providing that this dose does rodents—(1/10 the STD10; mg/m not cause severe, irreversible toxicity in the other mammalian (non-rodent) species tested (4). The starting dose for Phase 0 trials with a PK or PD endpoint is generally 1/50 the rat “no observed adverse effect level” (NOAEL). For studies that do not focus on a PD endpoint, the dose selected should allow a substantial margin of safety (e.g., a dose 100× as great did not cause toxicity in the single-dose toxicity study). For studies that address the mechanism of action, the FDA is willing to accept alternative approaches. Whereas a Phase I trial is designed to establish the MTD, the Phase 0 maximum dose can be that at which a PK/PD response is observed or target modulation is measured, providing that no drug-associated toxicity is found, and/or that the dose is less than 1/4 of the rat NOAEL, or that the total exposure to drug measured in human blood samples (area under the curve) approaches 1/2 of that measured in the most sensitive species (7).
Experience with the First Phase O Trial at the NCI
The first Phase 0 trial in oncology at the NCI was performed using ABT-888 (developed by Abbott Laboratories), an inhibitor of the DNA repair enzyme poly-ADP ribose polymerase (PARP) (10). Preclinical work by Abbott indicated that ABT-888 had favorable oral bioavailability characteristics and activity in human tumor xenograft models when combined (but alone alone) with a variety of chemotherapeutic agents that damage DNA (11). The NCI Phase 0 trial evaluated the effect of single 10, 25, or 50 mg oral doses of ABT-888 on PARP activity in tumor tissue and surrogate peripheral blood mononuclear cells (PBMC) in real-time. The study accrued fourteen patients, and no drug-associated toxicity was observed. The PD assay developed for this trial measured reduction in the amount of poly-ADP ribose (PAR) polymer as a surrogate for PARP inhibition. Using this assay, a statistically significant reduction in PAR was measured in both tumor biopsies and PBMCs (when using 25 mg or 50 mg doses). By correlating PD assay findings with the results from the PK analysis, a drug dose was determined that is likely to have target activity but that minimizes the risk of toxicity. These data were used to guide the design of several Phase I combination studies of ABT-888 with FDA-approved chemotherapeutic agents, each of which includes endpoints for PD as well as the safety and tolerability of the drug combination. The NCI Phase 0 trial met its primary objectives within five months and was completed before any of the planned Phase I studies were ready to start patient accrual.
PD Analysis and Drug Development
Critical for the success of a Phase 0 trial is a validated assay for experimental drug activity. The PARP assay for the NCI Phase 0 trial assay was validated using PD and PK data obtained from mouse xenograft tumor models to establish the optimal time window for drug administration and clinical sampling (12). Steps to consider when developing a PD assay are available on the NCI’s Developmental Therapeutics Program (DTP) Web site (13).
It is worth noting, however, that the presence of a validated assay is entirely distinct from having a validated biomarker. Validation demonstrates that the assay meets a set of design goals and performance criteria (Box 1). Generally, assay performance must be validated on each specimen type individually, a process that involves considerable investment of time and resources. The biomarker itself cannot be validated in advance because proving “fitness for purpose” requires a prospective clinical trial with an adequate number of patients. The biomarker can only be qualified to demonstrate that it gives the expected result in a set of model systems and reproducible values from the same set of specimens.
Suggested Performance Criteria for Pharmacodynamic Assay Validation
Accuracy
The ratio of the observed assay value to the actual quantity of analyte present.
Dynamic range
The concentration range over which the assay can accurately measure an analyte.
Precision
A measure of the variability of results around a determined value.
Reproducibility
The closeness of agreement between independent results obtained with the same methods and materials but under different conditions.
Sensitivity
Repeat determinations of the same specimen are possible.
The most desirable PD tests directly interrogate the status of a molecular target, but often this is not technically achievable. One of the issues arising in evaluating targeted cancer therapeutic agents is the inevitable shortcoming of using animal models to qualify data to support a certain mechanism of action and molecular target. A well-documented example of the complexity of this problem is the development of trastuzumab (Herceptin®). Experiments in severe combined immunodeficiency (SCID) mice demonstrated marked tumor responses (i.e., reduction in tumor mass) in xenografts that had increased HER2 expression, supporting a mechanism of action based on alteration of HER2 phosphorylation and receptor activity (14). However, intriguing results from non-immune-compromised models, and from patients, demonstrate that trastuzumab also kills tumor cells via antibody-dependent cellular cytotoxicity (ADCC) (15, 16). Which mechanism of action is more important in the clinic has not been conclusively demonstrated despite the success and widespread use of this drug.
An alternative to directly measuring drug mechanism of action is to develop surrogate markers of target function––the approach used in the NCI Phase 0 trial (10). The surrogate employed in the ABT-888 trial assessed changes in the amounts of enzyme products, assuming that there is a relationship between these products and changes in the activity of the enzyme itself, a generally accurate assumption. The ABT-888 PD assay examined cellular concentrations of PAR as a marker for PARP activity; decreased amounts of PAR following administration of a PARP inhibitor reflected PARP inhibition. However, the amount of product following an enzymatic reaction is not static. Such amounts are dynamically controlled by the balance between product synthesis and degradation. This important principle means that in tumor or blood samples with low rates of product degradation, even complete inhibition of product formation by the drug may not lead to a measurable change in the net amount of enzyme product. Tissues that contain insufficient activity to degrade enzyme products will yield false-negative PD results, even if the enzyme target of the drug is completely inhibited by therapy. Thus, the generation of false-negative PD results is a weakness of surrogate PD markers for target function and must be considered both when designing Phase 0 trials and interpreting study results. The small sample size of Phase 0 trials would generally not be sufficient to establish, with statistical certainty, whether a molecularly targeted anticancer agent has its anticipated effect, unless a dramatic effect of the drug is observed, as was the case for the ABT-888 study. Although more definitive PD studies require further evaluation in Phase I/II trials, Phase 0 studies provide an essential opportunity for the critical development and qualification of a PD marker under clinical conditions in patients before embarking on costly, later stages of drug development.
One unexpected finding from our assay development studies was the degree of heterogeneity of PAR amounts in tissue samples from mouse and human. Development of an assay with a dynamic range sufficient to differentiate inter- and intrapatient variability from drug treatment effects allowed us to examine differences in baseline PAR concentrations in humans. Earlier studies noted that amounts of PAR in peripheral blood mononuclear cells (PBMCs) of healthy volunteers changed considerably over time, but the significance of these values were obscured by lack of assay precision (17). Our observations confirm that baseline amounts of PAR in PBMC samples from healthy human volunteers vary widely, and that inter-day variation in patient PAR is also highly heterogeneous (18). Results from our PD validation studies indicated that up to 80% of the variability measured in samples from patients prior to treatment was random, which could have limited the possibility of establishing a correlation between tumor target and surrogate marker samples. Despite this potential for variability, we were fortunate to demonstrate an excellent correlation between the amounts of PAR in PBMCs and in tumor tissues in our trial (10).
Ethical Issues in the Conduct of Non-Therapeutic Clinical Trials
An area of concern during the planning for the development of cancer clinical trials under the FDA’s exploratory IND guidance is that the administration of sub-therapeutic doses of oncology agents confers no possibility of direct benefit to patients participating in Phase 0 trials. These patients are also obliged to give pre- and post-drug administration blood and biopsy samples for PD analysis. Participants are as carefully monitored for adverse effects as they would be on any other clinical trial, but, given that an Institutional Review Board (IRB) must conduct a risk:benefit assessment prior to protocol approval, we advocate consultation with and support from the institutional bioethics and human research committees prior to the initiation of Phase 0 clinical protocol development.
Participants in the NCI’s Phase 0 trials are highly motivated to take part in a research study that may benefit others. Most of these participants have been treated in other early clinical studies and are very well informed about the clinical trial options available to them both at the NCI and elsewhere (19). Also of importance is ensuring that patients who choose to participate in Phase 0 trials are not excluded from participating in future trials which do hold the possibility of therapeutic benefit (20). Patients who require immediate medical care for their cancer are excluded, but NCI Phase 0 inclusion criteria allow patients who are at least two weeks off previous therapy, or are waiting to enter another clinical trial, to participate. Other Phase I and II studies conducted at the NCI now allow patients to enroll within two weeks of completing participation in a Phase 0 trial.
Use of Phase O Trials to Combine Investigational Agents or for Molecular Imaging Studies
Two areas where Phase 0 trials may prove invaluable for drug development are studies combining molecularly targeted drugs and for clinical trials of molecular imaging agents. The opportunity to administer two or more experimental or FDA-approved drugs while collecting appropriate PK data will significantly enhance opportunities to understand the bioavailability of these agents. Correlating this information with PD data can provide evidence of any synergistic activity with minimal risk to patients from combination toxicity. Optimal relative doses and dosing schedules can therefore be determined prior to toxicology and tolerability assessment in Phase I testing.
Imaging studies with molecularly targeted agents have the potential to provide uniquely detailed information about drug distribution and target localization in vivo. Use of drug “microdoses,” which are defined in the exploratory IND as less than 1/100th of the dose required to produce a pharmacologic effect, to evaluate tissue distribution, measure tumor response after chemotherapy, or select patients for treatment, could inform many aspects of subsequent clinical evaluation (21, 22). The NCI is currently accruing patients to a Phase 0 trial of 111Indium-labeled trastuzumab in women with advanced stage breast cancer whose tumors express HER2/neu (23). The procedure involves first chelating the antibody and then linking the chelate to the 111Indium radionuclide. The resulting imaging agent has essentially the same affinity and activity as the therapeutic agent. The maximum dose of trastuzumab allowed in this study is 200 μg, considerably less than the recommended clinical dose of 2–4 mg/kg. Study objectives include correlating uptake of labeled antibody with the HER2 status of the tumor. Follow-up studies may involve chelating the antibody to particle-emitting radionuclides for targeted radiotherapy.
Conclusions
It will be some time before it will be known whether Phase 0 trials have had a positive impact on the development of new drugs for cancer or other indications. We anticipate that PD-driven studies will expedite the evaluation of those agents which directly modulate their targets. Our experience has been that conducting a Phase 0 trial does not delay clinical development or divert resources from Phase I investigations; rather, the reduced preclinical requirements for an exploratory IND enabled us to reach the Phase 0 study endpoints well before the first NCI-supported Phase I study of ABT-888 was ready to begin patient accrual. Furthermore, we were able to determine a dose of ABT-888 sufficient to cause maximum inhibition of PARP activity in vivo, sparing patient accrual to planned higher doses and the concomitant risk of drug-associated toxicity. Use of the lowest efficacious dose theoretically will allow an extra margin of safety for studies of combinations of ABT-888 with other agents, information that was applied during the design of Phase I trials of ABT-888 in combination with DNA damaging drugs.
It also remains to be seen whether the conduct of Phase 0 trials will result in more drugs “failing faster” than is currently the case. It is clear that the exploratory IND allows great flexibility in clinical development. Patient safety is still paramount, but the emphasis of Phase 0 first-in-human testing is on a drug’s target rather than its toxicity. Determining the MTD will still be required for further clinical development, but drugs that fail proof-of-principle target inhibition studies may be discarded before reaching formal Phase I/II evaluation.
Phase 0 trials are characterized by two critical determinants: 1) patients must be willing to participate in a clinical trial which offers no possibility of direct clinical benefit, and 2) serial blood (and often tumor) samples for research purposes are required. Our own experience has been that many patients with cancer are willing to participate in Phase 0 trials out of a sense of altruism and a desire to contribute to medical research. Although risks to the patient participating in a Phase 0 trial are considered low because of the limited drug exposure, they are not negligible. Every effort must still be made to minimize potential risk by selecting appropriate patients and monitoring their safety. Ethical considerations extend to ensuring that biopsy samples are only collected once the PD and PK parameters established during preclinical testing are met, and that the blood and tumor biopsy samples collected are put to the best possible research use.
The second critical determinant for Phase 0 trials is the integration of a robust and reliable assay of agent activity into the development plan. In addition to informing further clinical testing, the assay introduces the possibility of identifying biomarkers of drug efficacy in surrogate tissues. Validating the assay prior to clinical testing to measure whether the agent is or is not having its anticipated effect is labor- and resource-intensive, factors that may influence the decision to conduct a Phase 0 rather than Phase I trial. The availability of a qualified PD assay, however, is a requirement for measuring drug effect on target in tumor tissue. The development of the exploratory IND Guidance has provided researchers with the opportunity to evaluate molecularly-targeted agents in a context that facilitates clinical assay development; it is anticipated that this will lead to more rational and rapid evaluation of promising anticancer therapies.
Acknowledgments
We wish to thank Ms. Gina Uhlenbrauck, SAIC-Frederick, Inc., for editorial assistance in the preparation of this manuscript. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

James H. Doroshow, MD, received his medical degree, alpha omega alpha, from Harvard Medical School, Boston, Massachusetts, and performed his internal medicine residency training at the Massachusetts General Hospital, Boston. He spent three years at the National Cancer Institute (NCI) completing a fellowship in Medical Oncology. From 1983 to 2004, Dr. Doroshow was Chairman of the Department of Medical Oncology and Therapeutics Research and the Associate Director for Clinical Research at the City of Hope Comprehensive Cancer Center in Duarte, CA. Since 2004, he has been the Director of Division of Cancer Treatment and Diagnosis at NCI. He has authored over 300 publications in the fields of molecular and clinical pharmacology. Please address correspondence to JHD. Address correspondence to: Email: doro-shoj{at}mail.nih.gov; Fax: 301-496-0826.

Joseph E. Tomaszewski, MD, appointed as the Deputy Director of the Division of Cancer Treatment and Diagnosis in 2005, has been given the responsibility for developing a new pharmacodynamic initiative to support Phase 0 clinical trials under the FDA’s Exploratory-IND Guidance. For the last 16 years, he has served as the Chief, Toxicology & Pharmacology Branch in NCI with responsibility for the preclinical toxicological and pharmacological evaluation of most new cancer drugs developed by the NCI. This has resulted in the evaluation of over 180 diverse clinical candidates that has led to over 100 INDs and 5 NDAs.

Mel Simpson, PhD, heads the SAIC-Frederick protocol writing and support team in the NCI Division of Cancer Treatment and Diagnosis Project Management Office. She received her PhD in Biophysics from Kings College, London before conducting postdoctoral research in the Laboratory of Molecular Biology, National Institute of Diabetes and Digestive and Kidney Diseases, NIH. Dr. Simpson worked previously in regulatory consulting and academia on oncology and immunology drug evaluations.

Robert H. Wiltrout, PhD, leads the Center for Cancer Research (CCR), home to the basic, clinical, and translational research enterprise located within the National Cancer Institute, Bethesda and Frederick, Maryland. Dr. Wiltrout has authored more than 180 papers in the areas of innate immune response regulation and the biological therapy of renal cell carcinoma. He also contributes scientific and administrative leadership to the Center of Excellence in Immunology (CEI), which serves as a focal point for immunology research within CCR. He has been Visiting Professor of Immunology at the University of Rome and is a longstanding member of the American Association of Immunology, the American Association of Cancer Research, the Society for Leukocyte Biology, the Society for Natural Immunity, and the Society for Biological Therapy.

Lee J. Helman, MD, received his degree from the University of Maryland School of Medicine magna cum laude and was elected to Alpha Omega Alpha. He completed his internship and residency in Internal Medicine at Barnes Hospital Washington University. He began his fellowship training at the National Cancer Institute in 1983, where he has remained. He is Chief of Pediatric Oncology and Acting Scientific Director for Clinical Research, the Center for Cancer Research, National Cancer Institute. He is Professor of Pediatrics and Oncology at the Johns Hopkins University. He was elected to the American Society for Clinical Investigation and the American Association of Physicians.

Alice Chen, MD, is currently a Senior Investigator in the Targeted Therapeutics 1 Section in the Investigational Drug Branch of the Cancer Therapy Evaluation Program, NCI. Her fellowship in Medical Oncology was completed in 1991 at the Baylor College of Medicine in Houston, Texas. Dr. Chen then received additional training in drug development by completing the Oncology & Regulatory Sciences Fellowship at the FDA and NCI. She was a Research Officer through the Public Health Service at Bethesda Naval Hospital in the Navy Medical Oncology Branch from 1993–1998. Dr. Chen rejoined the NCI in 2005 and serves as a primary and associate investigator in a broad spectrum of clinical trials ranging from Phase 0 through Phase IV.

Melinda Hollingshead, DVM, PhD, was appointed Chief of the NCI Developmental Therapeutics Program’s Biological Testing Branch in 2005. With over 16 years experience in rodent models of cancer, she is responsible for generating preclinical efficacy data in support of compounds under development by the Division of Cancer Treatment and Diagnosis. She currently manages the preclinical rodent model component of the Phase 0 pharmacodynamic assay development effort. Dr. Hollingshead completed her veterinary medical degree at Auburn University and received a doctorate in immunology from North Carolina State University. Since then she has been involved with preclinical models for assessing potential chemotherapeutic agents for viral and neoplastic diseases. She has authored over 90 articles applying preclinical models to the drug development effort.

Sherry Yang, MD, PhD, is the Chief of National Clinical Target Validation Laboratory, Division of Cancer Treatment and Diagnosis, which develops and validates pharmacodynamic markers in Phase 0 and other phases of clinical trials. Her research focus has been on the clinical and/or translational research science, especially, on molecular target validation in clinical trials. Her recent publications include those on EGFR, HER2/neu, IGF1R, and VEGF receptor signaling pathway, tumor apoptosis pathway, multidrug resistance and epigenetic regulation of estrogen and progesterone receptors.

Seth M. Steinberg, PhD, received his BA in Mathematical Sciences from Johns Hopkins University, and his MS and PhD in Biostatistics from the University of North Carolina at Chapel Hill. Following three years as a statistician at the EMMES Corporation, he has been employed at the National Cancer Institute since 1986, initially as a staff fellow and now as a mathematical statistician and Head of the Biostatistics and Data Management Section of the Center for Cancer Research. His research interests primarily lie in the development and implementation of novel designs for early stage oncology clinical trials.

Oxana Pickeral, PhD, was Director, Strategic Alliance Management (contractor) at the Division of Cancer Treatment and Diagnosis, National Cancer Institute (NCI), NIH. She coordinated DCTD’s drug development partnerships within the Institute and with biopharmaceutical industry collaborators. She is also founder of an independent consultancy, BioVenture. Previously, she was a Bioinformatics Scientist at Human Genome Sciences, a Special Expert in bioinformatics at NCI, and trained at the NIH National Center for Biotechnology Information. She holds a PhD (Human Genetics) and an MBA from Johns Hopkins University, and did Master’s level studies in genetics at Brown University and St. Petersburg State University, Russia. She is currently with the Health Consulting Group of Booz-Allen, Inc.

Larry Rubinstein, PhD, was bestowed a doctoral degree from the University of Maryland, in statistics, has been a statistician in the Biometric Research Branch, NCI since 1985. He has co-authored methods for determining the required sample size in randomized studies, new designs for phase 1 trials, and appropriate designs for phase 2 trials. He participated in the development of the analysis tools currently used in the NCI in-vitro anticancer agent screen. He now does statistical review for 100–200 cancer clinical studies per year, as well as providing service on patient safety data monitoring committees, national cancer clinical trials steering committees, and statistical liaison with national cancer clinical trials organizations.

Jerry Collins, PhD, received his degree from the University of Pennsylvania and completed a postdoctoral fellowship at Johns Hopkins University School of Medicine. He spent a total of 11 years at the National Institutes of Health, including 5 years as Chief of the Pharmacokinetics Section at the National Cancer Institute. In 1988, Dr. Collins joined the FDA, where he directed the Clinical Pharmacology Lab for 17 years. In 2005, he returned to NIH to lead the Developmental Therapeutics Program at the National Cancer Institute. His 180 publications have emphasized PK/PD principles in oncology, especially for first-in-human studies.

Martin Gutierrez, MD, received his degree from the Javeriana University, Bogotá Colombia. He completed his training in internal medicine at Boston University and then served as chief resident in Medicine at the University Hospital, Boston University, Boston, MA. He received his training in Medical Oncology and Hematology at the US National Institutes of Health (NIH). Presently Dr. Gutierrez works at the US National Institutes of Health (NIH), where he has served as Head, Office of NCI-Navy Oncology and the Lung Cancer Clinical Research Section. His research interests focus on developmental therapeutics in cancer.

Anthony J. Murgo, MD, FACP, is Head of Early Clinical Trials Development at the National Cancer Institute Division of Treatment and Diagnosis, Adjunct Investigator and Head of the Developmental Therapeutics Section in the Center for Cancer Research Medical Oncology Branch, and Adjunct Professor of Medicine at the Uniformed Services University of the Health Sciences in Bethesda, MD. Since his training as a medical oncologist at Memorial Sloan-Kettering Cancer Center in New York City, Dr. Murgo has pursued a career in investigational cancer drug development with a current focus on early phase clinical trials.

Shivaani Kummar, MD, received her degree from Lady Hardinge Medical College, New Delhi, India, and performed her Internal Medicine Residency at Emory University, Atlanta, Georgia. Upon completion of her fellowship training in Medical Oncology and Hematology at the National Institutes of Health, Bethesda, MD, she joined Yale Cancer Center, Yale University, New Haven, Connecticut as an Assistant Professor in Medical Oncology. She worked closely with the Department of Pharmacology, Yale University to facilitate bringing new agents to clinical trials. At present, she is a Staff Clinician, Medical Oncology Branch, National Cancer Institute and works on early drug development in oncology.

Jay Ji, PhD, has been Laboratory Head and Senior Principal Scientist of the National Clinical Target Validation Lab at SAIC Frederick Inc., NCI-Frederick, since April 2006. He received his PhD in Genetics from Oregon State University and subsequently completed a postdoctoral fellowship in Pathology at the University of Washington. Dr. Ji was an NIH grantee from 1993 to 2003, first as a Senior Staff Fellow within the NIH intramural program and later as Director of Molecular Biology and Principal Investigator for NIH extramural programs. He also has over ten years experience in the in vitro diagnostic (IVD) industry directing biomarker discovery, assay development, and clinical testing programs. Dr. Ji has authored 30 research publications and has 4 patent applications in the fields of molecular diagnostics and biomarker validation.

Ralph E. Parchment, PhD, trained in Pharmacology & Experimental Therapeutics at the Johns Hopkins University School of Medicine and in Experimental Tumor Pathology at the University of Colorado. Previous positions included: Director, Pharmacology/ Toxicology Laboratory, Hipple Cancer Research Center (Dayton); Associate Professor, Internal Medicine and Pharmacology, Wayne State University; and Director, Pharmacology Core, Karmanos Cancer Institute (Detroit). He is the Director, Laboratory of Human Toxicology & Pharmacology, NCI-Frederick, where pharmacodynamic assays used in Phase 0 clinical trials are developed and validated, and clinical readiness proven. His research focuses on using in vitro assays of human drug response to predict clinical trial outcome.

Robert Kinders, PhD, is head of the Pharmacodynamics Assay Development laboratory at SAIC-Frederick, NCI-Frederick Cancer Research Facility. He received his degree in Biology at Kansas State University. His research has focused on cancer diagnostics and biomarkers. He has worked extensively in the U.S. Diagnostics Devices industry, including stints at Abbott Laboratories and C.R. Bard; and in pharmaceutical development at Abbott and CuraGen Corporation. He has served on the Technical Advisory Board at Abbott, and is currently on the Advisory Council of the Kansas Center for Basic Cancer Research.

