Emerging concepts from the recent literature
Sedate Subtypes of the GABAA Receptor
Anesthetic suppression of neuronal activity in the thalamus has been proposed as the basis for sedation and hypnosis produced by a wide variety of agents, including the prototypical inhaled anesthetic isoflurane. A variety of experimental approaches, including electrophysiological and brain imaging studies, corroborate an important role for the thalamus in sleep and wakefulness. But which of the many potential targets of isoflurane is actually responsible for quieting these neurons? In a new report from JPET, Jia and colleagues show that isoflurane enhances a tonically active inhibitory chloride current carried by α4 subunit–containing GABAA receptors and that this enhancement contributes to the ability of isoflurane to slow neuronal firing. The authors’ work points to further studies; for example, mice that lack the α4 subunit might be exploited to link this physiological effect of isoflurane to its behavioral actions. Understanding the molecular basis of anesthetic action has the potential not only to reveal how these drugs work, but also to contribute to our understanding of the neuronal basis of behavior. [Jia, F., Yue, M., Chandra, D., Homanics, G.E., Goldstein, P.A., and Harrison, N.L. Isoflurane is a potent modulator of extrasynaptic GABAA receptors in the thalamus. J. Pharmacol. Exp. Ther. Epub ahead of print; doi: 10.1124/jpet.107.134569 (2007).]—RA Pearce, University of Wisconsin.
Mrp2 Knockout Mice: Time for Model Makeovers?

The human ABCC2 (MRP2) transporter has a major effect on the absorption, elimination, and toxicity of many xenobiotics, and Mrp2 knockout mice have consequently been used to examine the role of this protein. Nevertheless, the mouse Mrp2 transporter has recently been shown to transport several clinically important compounds with efficiencies that vary markedly relative to the activity of the human ABCC2 ortholog. To address this concern, Zimmermann and colleagues have generated a number of cell lines that express either the mouse or the human transporter, revealing important differences between the the two ortholgs in the transport of various pharmacological agents. Moreover, the allosteric modulation of the ortholog transporters with regard to some drugs can be quite distinctive. As an example, sulfinpyrazone enhances the conjugate transport activity of the human protein but inhibits that of the mouse protein. Such disparities offer an important message of caution in the extrapolation of human drug effects and pharmacokinetics based on animal models. [Zimmermann, C., van de Wetering, K., van de Steeg, E. Wagenaar, E., Vens, C., and Schinkel, A.H. Species-dependent transport and modulation properties of human and mouse multidrug resistance protein 2 (MRP2/Mrp2, ABCC2/Abcc2). Drug Metab. Dispos. Epub ahead of print; doi: 10.1124/dmd.107.019620 (2008).]—B Sarkadi, Hungarian Academy of Sciences, Budapest
Glycinergic Tone: Dampening the Pain
Signals arising from painful peripheral stimulation are transmitted to the dorsal horn of the spinal cord via primary afferent neurons. Once entering the spinal cord, these signals can be positively or negatively modulated before ascending to supraspinal regions. Glycine is a unique transmitter in that it can both inhibit (acting at glycine receptors) or potentiate (acting at NMDA receptors) spinal nociceptive transmission. Using whole-cell electrophysiological recordings, Zhou et al. have investigated whether afferent input itself triggers glycinergic inhibitory potentials in spinal interneurons. The authors found that application of capsaicin, which activates nociceptive primary afferent neurons, enhanced glycinergic inhibitory potentials. These potentiated glycinergic potentials were blocked by application of ionotropic glutamate receptor antagonists. Thus, the release of glutamate into the dorsal horn following activation of nociceptive neurons can trigger the release of glycine to negatively regulate subsequent nociceptive transmissions. These findings point to an inherent endogenous mechanism that self-regulates the transmission of nociceptive signals in the spinal cord. [Zhou, H.-Y., Zhang, H.-M., Chen, S.-R., and Pan, H.-L. Increased C-fiber nociceptive input potentiates inhibitory glycinergic transmission in the spinal dorsal horn. J. Pharmacol. Exp. Ther. Epub ahead of print; doi:10.1124/jpet.107.133470]— TR Neelands, Abbott Laboratories
Binding Site for a HERG Channel Opener
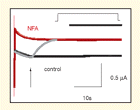
Familial mutations and drugs that affect HERG potassium channels can result in life-threatening arrhythmias. The development of drugs that can regulate HERG channels is thus of great interest, as is an understanding of the structural features that govern HERG function. Because different drugs have distinct electrophysiologic effects upon HERG channels, there are likely multiple binding sites that warrant investigation. Fernandez et al. now provide evidence that niflumic acid, a nonsteroidal antiinflammatory drug, binds to an extracellular site on HERG, identified by site-directed mutagenesis, and thereby modulates the voltage-dependence of channel activation. The authors’ study is novel and genuinely insightful, revealing a molecular binding pocket that mediates pharmacological shifts in the voltage dependence of a family of channels that has been implicated in important disease states and drug complications. [Fernandez, D., Sargent, J., Sachse, F.B., and Sanguinetti, M.C. Structural basis for ether-a-go-go–related gene K+ channel subtype–dependent activation by niflumic acid. Mol. Pharm. Epub ahead of print; doi: 10.1124/mol.107.043505 (2008).]—HJ Duff, University of Calgary, Canada
- © American Society for Pharmacology and Experimental Theraputics 2008

