Lipid Autacoids in Inflammation and Injury Responses: A Matter of Privilege
Abstract
Host defense is essential to all vertebrates, and programs of inflammation and wound healing must be highly integrated in tissue and organ structures, such as the skin, cornea, and mucosa, that provide crucial barriers and interfaces with the external environment. Certain aspects of inflammation and wound healing have posed a conundrum for biologists, especially to the extent that the two programs may appear to operate in opposition to each other. The recruitment of neutrophils to injured tissue, for example, is essential to inflammation and defense against infection, but can at the same time impair wound healing. One mechanism for regulating this duality is provided by lipid autacoids, which act to restrain leukocyte activation and to promote the resolution of inflammation. Emerging evidence indicates that lipid autacoids also have a central role in wound healing and in fact mediate a privileged injury response, as is observed in the cornea, characterized by rapid healing as well as effective host defense.
Introduction
Vertebrates have evolved remarkable mechanisms for the repair and maintenance of their own tissues (i.e., “host” tissues) that simultaneously preclude the invasion and growth of non-host cells and viruses. The front line of host defense relies on the skin, mucosal surfaces, and cornea, where epithelial tissues provide not only the critical physical barrier to a constant exposure to pathogens, but also an interface with commensal microbes (1, 2). Inflammation is a major component of host defense, and a fundamental feature of this vital response is the recruitment of leukocytes to sites of injury (3, 4). Polymorphonuclear leukocytes (PMN) and macrophages in particular are essential for preventing infection and the concomitant threat of life-threatening sepsis. Indeed, in humans, vulnerability to infection is an inevitable consequence of all known genetic or acquired defects in leukocyte function, including defects in adhesion, microbial killing, and phagocytosis; deficiencies in the generation of leukocytes in the bone marrow increase rates of infection and also other illnesses and raise mortality rates (1). In fact, any injury that compromises the external epithelial barrier triggers a robust inflammatory response.
Acute inflammation and wound healing are intimately linked responses that evolved to remove pathogens and noxious agents and ultimately restore tissue function and homeostasis. Acute wound healing and inflammation are tightly regulated responses that include highly complex programs with overlapping time course, common cell types, and shared chemical mediators (3–8). Delineation of these two vital injury responses has posed a major challenge, particularly in regard to a definitive role for inflammation and leukocytes in the wound healing response (5–7, 9, 10). Pharmacological suppression of the inflammatory response has become a major clinical target, primarily in an effort to control the precarious activation of powerful inflammatory responses that can involve “friendly fire” (e.g., leukocyte-mediated tissue injury), a key problem in inflammatory diseases and chronic wounds. Indeed, elegant studies employing knockout and knockdown approaches provide strong evidence that exacerbated inflammation impairs wound healing [see (5, 6, 9)]. Our prevailing paradigm thus suggests that nature tends to err on the site of caution, so that responses to injury can appear overzealous, triggering inflammation and impeding wound healing. On the other hand, several tissues—such as the oral mucosa and the cornea in particular—exhibit differential injury responses that are characterized by rapid wound healing and controlled inflammation, without compromising host defense (2, 9, 11, 14). A key feature of these privileged tissues upon injury is a restrained and self-resolving inflammatory response and the ability to control the precarious activation of PMN. A rapidly evolving field of research has begun to delineate endogenous circuits, depending on lipid autacoids, that restrain leukocyte responses and promote resolution of acute inflammation (3, 4, 15, 17). This review will focus on the emerging evidence that suggests that protective lipid autacoid circuits have a central role in privileged injury responses.
Leukocytes and Wound Healing
Recruited leukocytes are the predominant cell type at a site of epidermal/epithelial injury. They persist throughout the wound healing process and can reside within the tissue for days or weeks, even after successful wound healing (1), and their role in wound healing has been a major point of investigation (5, 6). During the acute inflammatory response, the delivery and removal of differing populations of leukocytes is orchestrated at injured tissue (4). An immediate response to cutaneous injury is the activation of platelets in injured blood vessels and their recruitment for the coagulation process. PMN are the first peripheral blood leukocytes to arrive at the site of injury, and their ability to remove bacteria, amplify inflammation, and release an arsenal of bactericidal agents establishes PMN as a primary effector cell in host defense. Macrophages represent the second wave of leukocyte effectors at the injured tissue, and they mark the transition of the inflammatory phase of the injury response to the proliferative phase, whereby they release an array of growth, angiogenic, and inflammatory factors. Moreover, macrophages have an important role in the removal of apoptotic PMN and, therefore, are pivotal to inflammatory resolution. Resident tissue mast cells and T lymphocytes appear after wound closure to regulate early and late phases of wound healing, respectively; the precise roles of these and other leucocytes remain to be defined (5–7).
Leukocytes (especially macrophages), platelets, and mast cells may with some justification be regarded as sources of growth factors (e.g., in angiogenesis) and may thus be presumed critical to the wound healing response. Indeed, the genetic deletion of either leukocyte-derived inflammatory mediators (e.g., MCP-1 and IL-6) or adhesion molecules (e.g., ICAM-1) impairs wound healing (5–7). At the same time, antibody knockdown approaches and more recent studies with knockout animals that lack macrophages and PMN (e.g., PU.1-null mice) provide strong evidence of functional redundancy among leukocytes, so that no single leukocyte type is required for proper healing of cutaneous wounds (5–7). Indeed, depletion of PMN in animal models significantly reduces scar formation and accelerates the rate of wound healing in the skin (5, 6, 9). The well-established concept of PMN-mediated tissue damage in ischemia-reperfusion injury and inflammatory diseases, along with the observed persistence of PMN in chronic wounds, condemns these cells as counterproductive to healing. This perception is reinforced by the remarkable ability of sterile fetal wounds to heal without forming scars, a process characterized by minimal inflammation and virtually no PMN infiltration; however, the amplification of pro-inflammatory networks—or genetic deletion of anti-inflammatory mediators (e.g., IL-10)—disrupt this privileged fetal reparative response and leads to scarring (5–7, 9). It is important to recognize that 1) these apparent contradictions arise from the artificial conditions of asepsis; that 2) all cutaneous or mucosal wounds are otherwise exposed to microbes; and 3) that the recruitment of PMN is essential to guard against infection. In this regard, it is striking that selected adult tissues, such as the oral mucosa and the eye, retain privileged injury responses while maintaining effective host defense. More importantly, these privileged injury responses are characterized by attenuated PMN recruitment and inflammation, which correlates with reduced scarring and rapid healing (2, 9, 11–14). Privileged adult tissues must thus have exceptional protective circuits, which balance host defense and healing by keeping the inflammatory response in check. These conserved circuits and their chemical mediators are of primary interest as they may lead us to therapeutic strategies for controlling inflammation and healing in privileged as well as non-privileged adult tissues.
Privileged Injury Response of the Cornea: A Perfect Balance?
The negative roles attributed to PMN and inflammation in wound healing present a conundrum; how has nature evolved a normal wound healing response without compromising vital host defense? The answer to this question appears to lie within endogenous circuits that regulate PMN activation and migration over the course of healing (3, 4, 15–17). The evolutionary requirement for a perfectly balanced inflammatory/reparative response is nowhere as critical and conserved as in the cornea (2, 11, 13). Under negative selective pressures, according to which vision was threatened by inflammation and scarring, this delicate, avascular tissue evolved a privileged injury response. The anterior surface of the eye, like mucosal surfaces, is constantly exposed to pathogens, antigens, and irritants. In addition, corneal epithelial cells are under constant sheer stress due to the regular blinking of eyelids at roughly six-second intervals. To add insult to injury, every night, during sleep, the avascular cornea is exposed to prolonged periods of hypoxia. While the eyelids remain closed during sleep, the cornea is in intimate contact with recruited PMN and inflammatory mediators, as evidenced by their abundant levels in the nocturnal tear film, which are rapidly removed upon the opening of the eyelids each morning. In short, the cornea successfully navigates through daily and lifelong cycles of prolonged hypoxia, subclinical inflammation, and PMN recruitment.
It stands to reason that the cornea must have developed remarkable endogenous circuits to control the precarious activation of PMN. This notion is strongly supported by the apparent paradox that normal wound healing in the cornea, after an abrasion injury, appears to depend on early PMN infiltration and pro-inflammatory mediators (14, 20–23). Several elegant studies employing antibody depletion of PMN and knockout mice that are deficient in adhesion molecules CD18, P- and E-selectin, and the leucine-rich proteoglycan lumican demonstrate that the attenuation of PMN infiltration and inflammation significantly delays re-epithelialization in the cornea (20–22). The cornea’s specialized protective circuits appear to provide a unique environment that unmasks a beneficial role of PMN in wound healing; however, there are limits to endogenous restraints over inflammatory responses. Even in the cornea, severe injuries, such as alkaline burn or infective wounds, trigger an exacerbated inflammatory response that clearly inhibits the normal wound healing (24, 25). The unique balance between acute inflammation and wound healing in response to epithelial injury in the cornea is dynamic. Identification of the central components of the cornea’s protective, anti-inflammatory circuits will require thoughtful probing of these circuits. One of the key regulatory components in these dynamic processes is mediated by lipid autacoids, and the following discussion will highlight recent evidence for their role in the wound healing and the privileged injury responses.
Lipid Autacoid Circuits
A diverse group of chemical mediators, including lipids, peptides, proteins, nucleotides, and bioactive gases orchestrate acute inflammatory/reparative responses. Lipid mediators such as eicosanoids, which are derived from the arachidonic acid (i.e., ω-6 C20:4), are among the earliest signals that are released in response to injury or an inflammatory stimulus (3, 16, 17, 26). Two families of enzymes, namely, the cyclooxygenases [i.e., cyclooxygenase-(COX)-1 and COX-2] and the lipoxygenases [including 5-lipoxygenase (5-LOX), 12-LOX, and 15-LOX], metabolize arachidonic acid to form lipid autacoids in humans (Figure 1⇓). It is widely appreciated that the COX-derived prostanoids (including thromboxane) and the LOX-derived leukotrienes and lipoxins play important roles as regulators of inflammatory and immune functions. These locally produced, short-lived lipid signals operate by engaging distinct classes of G protein–coupled receptors or nuclear receptors (17, 26, 27). In addition, arachidonic acid is a substrate of cytochrome P450 enzymes, which generate hydroxy and epoxy fatty acids with renal and vascular actions (28).
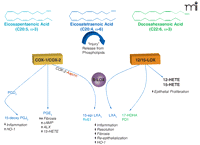
Formation of protective lipid autacoids in response to epithelial injury. Injury triggers phospholipase-dependent release of ω-6 (arachidonic acid) and ω-3 (EPA and DHA) from phospholipid pools. Arachidonic acid, EPA, and DHA are substrates in COX and LOX biosynthetic pathways that produce potent lipid autacoids that control inflammation and promote epithelial wound healing. These pathways include constitutive and inducible components that react to injury in the mucosa, epidermis, and cornea.
Arachidonic acid is not the only essential polyunsaturated fatty acid (PUFA) that is rapidly released by cells in response to stress, injury, or inflammatory stimuli. Several independent laboratories have demonstrated that the ω-3 PUFA eicosapentaenoic [EPA (i.e., ω-3, C20:5)] and docosahexaenoic acid [DHA (i.e., ω-3, C22:6)] are transformed, in a fashion analogous to eicosanoid metabolism, by COX-2 and LOX enzymes (Figure 1⇑) to generate novel classes of endogenous lipid autacoids with anti-inflammatory and protective activities (29, 30). The physiological and developmental roles of essential ω-6 and ω-3 PUFAs, such as arachidonic acid (ω-6) and DHA (ω-3), along with their dietary metabolic precursors linoleic acid (ω-6, C18:2) and alpha linolenic acid (ω-3, C18:3), are well documented (31, 32). More importantly, virtually every cell expresses at least one enzyme (and usually multiple enzymes) to produce potent mediators of inflammation derived from PUFAs. Hence, it is not surprising that several independent lines of evidence demonstrate an indirect role for lipid autacoids in the wound healing response (see below).
A paradigm shift in our appreciation of eicosanoid activity in inflammation occurred with the revelation that eicosanoids not only initiate, amplify, and perpetuate inflammation, but they also act as key regulators that promote the resolution of inflammatory responses. Several elegant reviews have recently defined the resolution of inflammation as a biologically programmed process that is quite distinct from the mere suppression of inflammation (3, 4, 33). The 1984 discovery of the lipoxins as a new class of eicosanoids championed the concept of anti-inflammatory and pro-resolving lipid autacoids and spurred the discovery of a growing family of anti-inflammatory lipid mediators generated via COX-2 and/or LOX pathways (Figure 1⇑). Among the protective lipid autacoids that have been elucidated are lipoxin A4 (LXA4), 15-deoxy-prostaglandin J2 (15-deoxy PGJ2), EPA-derived resolvin E1 (RvE1), and the DHA-derived resolvins (RvD1) and protectin D1 (PD1) (3, 4, 15, 17, 29, 30, 33–35). Indeed, a recent seminal study underscores the important role of COX-2 and LOX enzymes in the temporal generation of inflammatory and pro-resolving mediators (36). Inhibition of either enzyme activity results in deficient removal of apoptotic PMN from sites of acute inflammation. More importantly, several structurally distinct endogenous lipid autacoids, namely, LXA4 (37), RvE1, and PD1 (36), not only demonstrate anti-inflammatory activities, but also can stimulate macrophage phagocytosis of apoptotic PMN, an essential step in resolving acute inflammation.
Cyclooxygenases and Wound Healing
Inhibition of prostaglandin activity is one of the most widely used clinical strategies to combat inflammation and inflammatory pain, and no other lipid autacoid pathway has been as extensively studied in terms of biosynthesis, bioactivity, and receptor signaling (17, 38). Despite an impressive body of work concerning the role of prostaglandins in wound healing, however, their role as key regulators of inflammation remains to be fully defined (6, 7). A central role for the COX pathways has been demonstrated in the repair of bone fractures and the healing of gastric and colonic ulcers.
A striking feature of epithelial and bone injury is the fact that the earliest responses to injury includes induction of the early-response gene that encodes COX-2 and the formation of prostaglandins. Prostaglandins manifest multiple activities, and the temporal and spatial contexts in which they operate determine their biological roles, which often can oppose each other. For example, the COX pathway has well-established roles in regulating bone biology, osteoclastogenesis, bone formation, and fracture repair (39). Prostaglandins can thus influence bone resorption, which contributes to the pathogenesis of localized aggressive periodontitis (40); on the other hand, prostaglandins are critical to bone formation, an essential component of fracture repair (39). Interestingly, both of these activities appear to be mediated by prostaglandin E2 (PGE2), which had previously been regarded predominantly as an inflammatory mediator.
The early-response gene that encodes COX-2 is also highly expressed in response to mucosal injury and ulcers. Inhibition of COX, especially of COX-2, by nonsteroidal anti-inflammatory drugs (NSAIDS) significantly exacerbates mucosal injury induced by inflammation and impairs wound healing of gastric and colonic ulcers (41, 42), findings that further undermine the dogma that had strictly cast COX-2 with a role in the initiation and amplification of inflammation. We now appreciate the protective roles of COX-2 against mucosal injury, attributed to the formation of anti-inflammatory and pro-resolving lipid autacoids (Figure 1⇑) such as 15-epi-LXA4, an LXA4 isomer whose formation can be initiated by aspirin-acetylated COX-2, and 15-deoxy-PGJ2 (17, 42). Moreover, PGE2, a prominent product of the COX-2 pathway, plays a pivotal role in checking leukocyte function by activating specific PGE2 receptor EP2 and EP4 and thereby increase intracellular cAMP levels.. Activation of the cAMP-protein kinase A signaling cascade switches the eicosanoid profiles of PMN, so that production of the pro-inflammatory leukotriene B4 is replaced by production of the anti-inflammatory LXA4 (43). PGE2 can also upregulate the expression of the LXA4 receptor in epithelial cells, which underscores the pleiotropic actions of this prominent prostaglandin in inflammation.
COX-2 induction is one of the earliest responses to cutaneous injury, and the sequential deployment of pro- and anti-inflammatory prostaglandin signaling correlates with the progression of the healing response (6, 7, 45). However, data from COX-2 and COX-1 knockout mice and experiments with pharmacological inhibitors suggest that neither isozyme is essential for normal cutaneous wound healing. Nevertheless, PGE2 is a primary product of the epidermis, clearly regulates fibroblast migration and contraction, and depending on the tissue either promotes (skin) or inhibits (lung) the fibrotic process (6, 7, 46–49). Moreover, impaired wound healing in the diabetic ob/ob mouse is associated with dysregulation of COX-1 and impaired formation of PGE2/PGD2 (50). Thus, although there is likely some redundancy of pathways, normal cutaneous wound healing in the epidermis is likely dependent on COX signaling, especially when wound healing is dysregulated.
Lipoxygenases and Wound Healing
The major eicosanoids generated by human mucosal epithelial cells and the epidermis are mono-hydroxy fatty acids, especially 15-hydroxy eicosatetraenoic acid (15-HETE). It is well established that PUFAs are essential to the normal physiology and barrier function of skin. Moreover, the observation that the skin has a high rate of PUFA metabolism, along with the fact that 12-LOX and 15-LOX are highly expressed in the epidermis and epithelial cells, strongly supports a central role for LOX pathways in normal skin physiology (7, 26, 51, 52). Indeed, genetic deletion of the platelet type 12-LOX in mice increases transepidermal water loss, and the primary 12-LOX metabolite, 12-HETE, is implicated in human inflammatory diseases of the skin such as psoriasis (53–55). Moreover, linoleic acid and dihomo-γ-linolenic acid can be metabolized by 15-LOX to generate several hydroxy fatty acids, which may have a role in proliferation and inflammation of the epidermis (56). Unlike their well-established roles in generating potent mediators of inflammatory and immune responses, however, the functions of LOX enzymes in wound healing are not well defined (7, 26, 51, 57).
An enzyme of particular interest is human 15-LOX (ALOX15) as it is considered one of the most prominent inducible genes in monocytes; it is also highly expressed in human mucosal and corneal epithelial cells (14, 52, 57, 57). More importantly, 15-LOX is a key enzyme in mucosal tissues for the formation of LXA4 and DHA-derived resolvins and protectins (29, 59). These prominent anti-inflammatory and pro-resolving autacoids restrain PMN and lymphocyte activation, dendritic cell function, formation of pro-inflammatory chemokines, and promote clearance of apoptotic PMN by macrophages (3, 15, 27, 29,30, 33). These oxygenated fatty acids are highly conserved among vertebrates, including fish and frogs, and human pathogens, such as Pseudomonas areuginosa and Toxoplasma gondii, exploit this critical anti-inflammatory lipid circuit to suppress host immune responses (60, 61).
A striking feature in both mouse and human corneas, both of which exhibit privileged injury responses, is the high epithelial expression of Alox15 (cf. 12/15-LOX) and 15-LOX (ALOX15 and ALOX15B), respectively (14, 58, 62). Our laboratory recently reported on the epithelial dependent formation of LXA4 and expression of the LXA4 receptor (ALX) in the cornea (14, 23), thereby assigning a function to this prominent enzyme in the cornea. Specifically, we found that Alox15 knockout mice provide a loss-of-function model (25) in which LXA4 levels are reduced and epithelial wound healing is delayed. Topical treatment with LXA4, 17S-hydroxy DHA, and PD1 restored wound healing, a response that is distinct from the anti-inflammatory and leukocyte-specific activities of these lipid derivatives in the cornea (14, 23, 25).
The molecular mechanisms that underlie the epithelial-specific actions of 15-LOX-derived lipid autacoids, which likely act as second messengers and/or receptor agonists, are just beginning to be elucidated (Figure 2⇓). Key observations come from expression studies of the LXA4 receptor in corneal and mucosal epithelial cells (14, 27, 44, 63) and the identification of 15-HETE as a second messenger in epidermal growth factor-(EGF)– and hepatocyte growth factor-(HGF)–mediated proliferation of human corneal epithelial cells (64). Moreover, changes in the expression of human 15-LOX-1 and 15-LOX-2 correlate with colonic and prostate epithelial differentiation/proliferation.
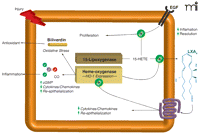
Interaction of protective circuits in the privileged injury response. Lipoxygenase (15-LOX) and heme–oxygenase (HO) pathways operate in corneal epithelial cells to generate autacoids that act in both intracellular and extracellular capacities. Carbon monoxide and LXA4 attenuate the expression of epithelial-derived pro-inflammatory mediators. Biliverdin is a potent antioxidant, and 15-HETE serves as a second messenger for epithelial growth factor (EGF). The HO system promotes formation of LXA4 in the cornea, and activation of the LXA4 receptor in turn amplifies expression of HO-1. Released autacoids from both pathways limit leukocyte function (PMN). A potential positive feedback loop is thus elaborated to elevate anti-inflammatory and cytoprotective autacoids to control the precarious activation of the inflammatory response.
Lipid Autacoid Interactions with Cytoprotective Heme Oxygenase
Most adult tissues express 15-LOX and generate protective lipid autacoids, but they do not exhibit a privileged injury response. So, what sets the cornea apart from the typical adult tissue? A potential explanation is an elevated basal tone of protective mediators, which would raise the threshold for shifting inflammation from a restrained to an exacerbated response. We recently identified the interaction of two prominent protective circuits in the cornea that may operate to amplify the anti-inflammatory tone of the immune-privileged cornea (25, 65).
In addition to expressing 15-LOX, the cornea also expresses a cytoprotective heme oxygenase (HO) system (25, 65, 66). Heme oxygenases (HO-1 and HO-2) are essential cytoprotective activities that metabolize heme and generate antioxidants and the bioactive gas carbon monoxide (67–70). HO-1 has emerged as an inducible and essential cytoprotective agent, and heightened expression of HO-1 results in striking anti-proliferative, anti-inflammatory, and anti-apoptotic actions; HO-2 is constitutively expressed in normal skin and cornea. It is thus of particular interest that epithelial abrasion in the cornea, which is normally characterized by self-resolving inflammation and complete wound healing within five to seven days, can elicit chronic inflammation without wound healing if constitutive expression of the cytoprotective HO system is disrupted (25, 65).
The HO system generates two prominent protective mediators (67–70): 1) the anti-oxidant bilirubin; and 2) carbon monoxide. Carbon monoxide is a potent activator of guanylate cyclase and regulates MAP kinase pathways to result in lower levels of the pro-inflammatory cytokines IL-6, TNFα, IL-1β, and MIP-1α. Significantly, anti-inflammatory lipid autacoids such as LXA4, PD1, and RvE1, downregulate the same profile of pro-inflammatory cytokines (27, 29) that is targeted in carbon monoxide–mediated cytoprotection. More importantly, several independent laboratories have demonstrated that anti-inflammatory lipid autacoids, namely, 15-deoxy-PGJ2, LXA4 and PD1, can induce or amplify the expression of the cytoprotective HO-1 system (25, 71, 72). The anti-inflammatory lipid autacoids and the HO system appear to interact and complement one another. We have employed a loss-of-function approach using HO-2 and 12/15-LOX knockout mouse strains, both of which manifest impaired wound healing and exacerbated inflammation in response to epithelial injury (14, 23, 25, 65). Intriguingly, the absence either of the two protective pathways impinges on the expression and/or function of the second protective pathway in the cornea. Specifically, mice deficient in 12/15-LOX do not invoke a robust HO-1 system–mediated response to an abrasion injury unless they are treated with topical LXA4. Correspondingly, mice deficient in the HO system, with a phenotype of chronic inflammation and failed wound healing, demonstrate a ~50% reduction in endogenous LXA4 formation.
Taken together, these findings suggest that the two prominent anti-inflammatory systems that are resident in the cornea interact significantly and may together represent a positive amplification loop to maintain elevated tone of anti-inflammatory signals (Figure 2⇑). It remains to be determined if this interdependence is present in other tissues or is unique to the privileged injury response of the cornea. Recent reports have demonstrated that systemic treatment with LXA4 or PD1 amplifies renal HO-1 expression (73), which is associated with attenuated inflammation, renoprotection, and reduced interstitial fibrosis after ischemic renal injury (74). It is thus tempting to speculate that the endogenous HO-1/15-LOX axis might be clinically exploited to keep inflammation in check while promoting normal wound healing.
Conclusion
Host defense responses to epithelial and epidermal injury appear to rely on redundant circuits that tightly control the precarious, but essential, activation of the inflammatory processes. Anti-inflammatory lipid autacoids, such 15-LOX–derived LXA4 and PD1 and COX-derived 15-deoxy PGJ2, have emerged as central regulators of leukocyte function and the active resolution of inflammation. Biosynthetic pathways and receptors for these lipid autacoids are highly expressed in the mucosa, epidermis, and cornea. In privileged tissues such as the cornea, anti-inflammatory lipid autacoids provide a wide spectrum of protective actions, which include: 1) control of leukocyte activation; 2) active resolution of PMN activities; 3) amplification of the resident cytoprotective heme–oxygenase system; 4) attenuation of pro-inflammatory mediators; 5) proliferative effects on epithelial cells; and 6) re-epithelialization of injured tissues. Their roles in inflammation and wound healing provide a compelling argument for the investigation of lipid autacoids as therapeutic targets and agents.
Acknowledgments
I gratefully acknowledge the National Institutes of Health for supporting research in the author’s laboratory (EY016136 and HL34300), which is cited in this review.
- © American Society for Pharmacology and Experimental Theraputics 2008
References
Karsten Gronert, PhD, is Acting Associate Professor and Solon M. and Pearl A. Braff Chair in Clinical Optometric Science at the University of California at Berkely School of Optometry. His research centers on the protective circuits that regulate essential inflammatory/reparative responses and relates to new therapeutic approaches to minimize and control inflammatory damage and diseases. E-mail kgronert{at}berkeley.edu; fax 510-643-5109.

