Taste Cells of the Gut and Gastrointestinal Chemosensation
Humans consciously perceive only five distinct tastes: bitter, sweet, umami [the savory taste of monosodium glutamate (MSG)], salty, and sour. Taste cells of the tongue utilize G protein–coupled receptors (GPCRs), the heterotrimeric G-protein gustducin, and downstream signaling cascades to detect bitter, sweet, and umami compounds. The mechanisms of salty and sour taste transduction are less certain but may utilize the epithelial sodium channel (ENaC), polycystic kidney disease (PKD) channels, transient receptor potential channel M5 (Trpm5), or other ion channels. Sugars, such as glucose and fructose, and non-caloric sweeteners, such as sucralose, activate the tongue’s heterodimeric T1R2+T1R3 sweet receptor, whereas MSG and other savory amino acids are detected by the T1R1+T1R3 heterodimer. Toxic plant alkaloids, such as strychnine and other bitter compounds, activate one or more of approximately thirty divergent T2R bitter receptors.
A number of researchers had speculated that chemosensory detection of glucose and other macronutrients in the lumen of the gut might utilize cells and receptors like those of the tongue (1). Indeed, in 1996 Höfer et al. showed that the taste cell–specific G-protein gustducin is also present in the gut of rodents (2). Subsequent studies during the past few years have shown that T1R sweet and umami taste receptors and T2R bitter taste receptors are present in the guts of rodents and humans. Only within the last year, however, have the functions in gut of these taste signaling elements been uncovered (3, 4). The gut “tastes” sugars and sweeteners in much the same way as does the tongue and by using many of the same signaling elements.
It has long been known that orally ingested glucose is much more effective than intravenous glucose in raising plasma insulin concentrations (this is called the incretin effect) (5) (Box 1). Glucose in the gut lumen leads to secretion, in a concentration-dependent manner, of incretins [the gut hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP, also termed gastric inhibitory peptide)], which in turn augment insulin release from the pancreas. Yet, how glucose in the gut lumen stimulates release of incretins was not known until 2007: glucose in the gut activates sweet taste receptors and gustducin present in the intestine’s enteroendocrine L cells, leading to release of GLP-1 from these cells. Gustducin is also found in another subset of enteroendocrine cells—the K cells—which express GIP in response to food intake. A third set of enteroendocrine cells, the L/K cells, express both GLP-1 and GIP along with gustducin. GLP-1, GIP, and other gut hormones released from the K and L cells contribute to a wide variety of functions, including gut motility, nutrient absorption, insulin secretion and glucose homeostasis, whole body energy assimilation, satiety–hunger signaling, and cellular proliferation (6).
Incretins
The process of digesting food elicits release of hormones, termed incretins, from specialized endocrine cells of the gastrointestinal tract. Incretins positively modulate the amount of insulin released from the beta cells of the islets of Langerhans in the pancreas in response to rising blood glucose. Incretins also slow down the rate of gastric emptying and gut motility and have other effects outside of the pancreas that influence glucose homeostasis. Glucagon-like peptide-1 (GLP-1) and gastric inhibitory peptide (GIP) are incretins whose activities are neutralized by dipeptidyl peptidase 4 (DPP 4) present on endothelial cells in blood vessels, on lymphocytes, and in the circulation. An agonist of the GLP-1 receptor, called exenatide, that is not subject to DPP 4 degradation, is now available to treat type 2 diabetes, as is sitagliptin, a DPP 4 inhibitor.
Gustducin and taste-signaling elements are expressed selectively in K and L, and perhaps other, enteroendocrine cells but not in the more numerous enterocytes lining the gut that are directly involved in absorbing sugars and other nutrients released from digested food. However, the gustducin- and sweet receptor-expressing enteroendocrine cells appear to regulate glucose transport into enterocytes. Glucose and galactose enter enterocytes through the Na+, glucose cotransporter 1 (SGLT1), whereas fructose is absorbed through another apical transporter, GLUT5. The sugars then passively diffuse from the enterocytes into the bloodstream through GLUT2 in the enterocytes’ basal membranes. The amount of expression of SGLT1 in enterocytes is regulated by diet: high carbohydrate diets or supplementation with sugar- or sucralose-sweetened drinking water elevate SGLT1 expression and glucose-uptake capacity (4). Mice genetically modified to lack gustducin or T1R3 fail to increase SGLT1 expression or glucose uptake on a high sugar diet or when fed artificial sweeteners like sucralose, implicating these taste signaling elements in regulation of SGLT1. Glucose concentrations greater than 30 mM saturate SGLT1 transport capacity, and humans regularly consume more sugar over short time frames, such as in the 75 gram oral glucose tolerance test (OGTT) or the 40 grams of sugar in cans of regular soft-drink than is possible to transport through SGLT1. Under those conditions, there is, within minutes, a large increase in the presence of GLUT2 in brush border membranes (BBM), thereby providing increased sugar diffusion capacity down a concentration gradient from the lumen into the enterocytes to match dietary intake. Indeed, gut-expressed taste receptors have been implicated in sugar-induced GLUT2 trafficking (7). Sugar in the diet stimulates taste signaling elements on enteroendocrine cells to promote release of their hormones—in turn, these hormones regulate the expression of SGLT1 and GLUT2 in enterocyte BBM (4, 8) (Figure 1⇓).
T2R bitter taste receptors also have been found in gut (9, 10). Bitter agonists elicit cholecystokinin (CCK) secretion from enteroendocrine STC-1 cells and increase intracellular calcium concentrations in HuTu-80 and NCI-H716 cells (10, 11). Additonally, bitter compounds placed directly into rat stomach delay gastric emptying and elicit aversion to a flavored drink (12). The L-cell hormone peptide YY (PYY) evokes an aversive response to food (13), whereas GLP-1 delays gastric emptying and decreases food intake. Thus, there is the distinct possibility that bitter compound–mediated activation of gut-expressed T2Rs could promote protective physiological responses through release of the enteroendocrine hormones so as to lessen the arrival of possibly toxic (usually bitter tasting) agents into the gut.
Are taste receptors in gut relevant to diet-related disease and other public health issues? Could they serve as novel targets for new therapeutics? As is well known, obesity in the United States is rampant. In addition, the prevalence of the metabolic syndrome, a cluster of cardiovascular risk factors associated with type II diabetes, hyperlipidemia, and high blood pressure, is also increasing as a continuous variable (14, 15). Increased obesity and hyperlipidemia have been linked to increased consumption of sugary soft drinks (16–18); however, diet soft-drinks containing non-caloric sweeteners may further increase the incidence of obesity and/or metabolic syndrome (19–21). A rising prevalence of metabolic syndrome was found with increasing numbers of soft-drinks consumed: furthermore, the prevalence was even slightly higher, can-for-can, in the diet soft-drink drinkers vs regular soft-drink drinkers. Lutsey et al. found that the risk of developing metabolic syndrome was 34% higher in those drinking the most soft-drinks compared to those drinking the fewest. Other smaller studies have also reported associations of diet soft drinks with high blood pressure in women and weight gain in boys (22, 23). Is it something in the diet soda or is it a predisposing behavior of those consuming diet soft-drinks (e.g., unhealthy eating habits) that leads to increasing weight gain and metabolic syndrome? Rodent data suggest that sweeteners per se alter feeding behavior. Rats given low-energy saccharin-flavored gelatin overeat and become obese, relative to rats fed high-energy (starch) saccharin-flavored food (24). In a very recent study, rats receiving saccharin-sweetened yogurt gained more weight due to increased food intake than rats given sugar-sweetened yogurt (25). In light of all this human and animal data, caution is warranted against the television advertisements advocating addition of artificial sweetener instead of sugar to morning cereals for children.
Might the effects of non-caloric sweetener ingestion on metabolic syndrome and obesity be mediated in part by gut taste receptors? Non-caloric sweeteners do have many of the same effects on gut taste receptors as do energy-containing sugars: they each induce hormone release, increase SGLT1 expression, and induce GLUT2 translocation to the BBM (3, 4, 7). Ingestion of non-caloric sweeteners may prepare the gut for the presence of nutrients in the same manner as the nutrients themselves, but in the absence of a source of calories the balance between taste receptor activation, nutrient assimilation and appetite may be disequilibrated leading to an increase in appetite and overeating of unnecessary calories when they are readily available. Could it be that gut-bypass surgery leads to diminished appetite by bypassing the gut’s taste receptors? Two novel non-surgical therapeutic approaches come to mind. First, diabetes might be treated by activating the taste receptors on gut L cells so that they release GLP-1 to augment insulin release. Second, compounds that block activation of the gut’s taste receptors might serve as appetite suppressants. The potential of these approaches could readily be evaluated in animal studies using enteric-released sweeteners (e.g. sucralose) as activators and enteric-released sweetener antagonists (e.g. lactisole) as inhibitors of L cell taste receptors to, respectively, promote or inhibit release of GLP-1. Whereas a spoonful of sugar may act on the tongue’s taste buds to help the medicine go down, the actions of sweeteners and sweet antagonists in the gut may be therapeutic in their own right.
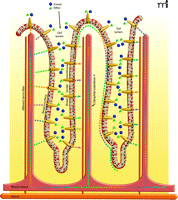
Physiological consequences of activating gut taste cells. Enteroendocrine cells (orange comet-shaped cells) in gut villi have functional sweet and bitter taste receptors (not shown) at their apical (luminal) membranes. Sugars and other nutrients within the gut lumen activate the gut taste receptors leading to release of enteroendocrine cell hormones. The incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP), as well as other hormones (colored dots), are released from the basal surface of the enteroendocrine cells into the interstitial space, where they may act locally as paracrine factors, neurotransmitters and neuromodulators, or enter the blood stream and act as classical hormones at distal sites. Locally, the incretins and other enteroendocrine hormones elicit increased expression of the SGLT1 transporter and translocation of the GLUT2 transporter to the brush-border surface of the enterocytes, resulting in rapid absorption of glucose, galactose, and fructose. In addition, incretins activate afferent neurons in villi, sending signals to the central nervous system and influencing activity of the enteric nervous system. The enzyme dipeptidyl peptidase 4, present on epithelial cells of blood vessels, including those in the gut interstitium, and in the circulation, quickly degrades the biologically active forms of circulating incretins. Locally active incretins that are subject to less-rapid degradation may have a more prolonged duration of action.
Acknowledgments
The authors are supported by the Intramural Research Program of the NIA/NIH (JME) and grants from the NIH/NIDCD (RFM). The views expressed here are the authors’ own and do not reflect those of the NIH or DHHS. RFM has a personal financial interest in the form of stock ownership in the Redpoint Bio company, receives consulting fees from Redpoint Bio, and is an inventor on patents and patent applications which have been licensed to Redpoint Bio.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Robert F. Margolskee, MD, PhD, is Professor in the Department of Neuroscience, at the Mount Sinai School of Medicine. His research is focused on molecular mechanisms of taste signaling in tongue and chemosensation in gut. E-mail robert.margolskee{at}mssm.edu; fax 212-849-2599.

Josephine M. Egan, MD, is an Endocrinologist and chief of the Diabetes Section, in the Intramural program of the NIA/NIH. She studies incretins, specifically their mechanisms of release, mechanisms of action, and their role(s) in diabetes.

