Schizophrenia
Moving Beyond Monoamine Antagonists
Abstract
Schizophrenia is a disabling psychiatric disorder characterized by positive, negative, and cognitive symptoms. The first pharmacological treatments for schizophrenia were discovered by serendipitous, albeit carefully documented, clinical observations. The discovery of chlorpromazine and other dopamine D2 receptor antagonists as antipsychotic agents set the early course of drug discovery in the context of schizophrenia and other psychiatric disorders, and various monoamine receptors became the prime focus of neuropharmacological studies. Success in treating the positive symptoms nevertheless remained limited by the general lack of efficacy in addressing negative symptoms and cognitive impairment. In recent years, several new experimental approaches have emerged for the identification and treatment of different symptom clusters that do not rely on blockade of monoamine receptors. Muscarinic, nicotinic, and glutamatergic signaling mechanisms have become essential to neuropharmacological and behavioral models of discrete aspects of schizophrenia. And as a consequence of these insights, novel drug entities have become available to study and potentially treat the disabling cognitive and negative symptoms of psychiatric disease. Current attempts to target a new range of receptors entail unprecedented fine-tuning in the pharmacological manipulation of specific receptor subtypes.

Introduction1
Schizophrenia is a chronic psychotic disorder, the pathophysiology, etiology, and rational treatment of which remain uncertain. It was defined as a disease entity by Kraepelin around 1890, who distinguished it from bipolar disorder by symptoms and outcome; however, historical records document the illness, variously named, throughout history. Throughout the twentieth century, schizophrenia was conceptualized as a unitary illness, with a sole explanatory pathophysiology and treatment target. Antipsychotic treatments (first- and second-generation drugs) conformed to this formulation in that they were all antagonists of dopamine (D2), serotonin (5HT2A), and other monoamine receptors. The conceptualization of treatment targets, however, has progressed, even without a defining biology. The current formulation of disease manifestations proposes that there are discrete but overlapping “component symptom complexes” that make up the illness; they are phenomenologically distinct, independent in course and onset, and pharmacologically distinguishable. Treatment targets represent component complexes to the extent that they can be monitored for clinical response (1).
The symptoms of schizophrenia cluster into several overlapping sets, including positive, cognitive and negative symptoms (Figure 1⇓). The most prominent are the positive psychotic symptoms, including hallucinations, delusions and thought disorder. This symptom set typically appears in late adolescence or early adulthood and is characteristically episodic; it is most virulent during adult years, although it attenuates in some patients as they reach their 50s. Psychotic symptoms continue to define diagnosis.
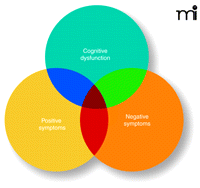
The complexity of symptoms in schizophrenia. No longer seen as a unitary illness, a variety of molecular pathways can be engaged in the disorder, and symptoms manifest with great variability among patients. Current research into pharmacological targets seeks to address the complexity in terms of symptom sets and receptor subtypes.
Cognitive dysfunction, another symptom complex, includes predominantly deficits in attention, executive dysfunction, and memory. These symptoms are often present before the onset of illness (i.e., prior to florid psychosis) and can be seen in asymptomatic family members. Cognitive symptoms are stable throughout life and are not responsive to antipsychotic drugs.
Negative symptoms are represented by anhedonia, alogia, and asociality. These characteristics most resemble cognitive dysfunction, and negative symptoms may in fact be related to cognitive symptoms. Symptom complexes are highly variable from person to person, so that each patient presents with an individual mix of symptom sets.
Novel Approaches to the Treatment of Cognition in Schizophrenia
In optimally treated people with schizophrenia, poor psychosocial function correlates not with residual psychotic symptoms, but rather with cognitive dysfunction (2). Poor executive function predicts low overall community function, low vigilance, poor social skills, memory deficits, and poor overall outcome. Indeed, it has become clear, just focusing on positive symptoms, that satisfactory treatment exists for psychosis. It is increasingly recognized that cognition, on the other hand, is not appreciably affected by antipsychotic medication, and this observation has redirected the focus of experimental therapeutics on the discovery of treatment targets and compounds to augment cognitive function in the illness. The MATRICS initiative (Measurement and Treatment Research to Improve Cognition in Schizophrenia; Figure 2⇓), defines this new focus. This NIMH-sponsored initiative was established to describe the nature of the cognition deficit in schizophrenia (3), to judge putative molecular targets for cognition (4), to develop a broad rating instrument to quantify cognitive dysfunction, and to design clinical trials (5). MATRICS has thus sought to accelerate the identification and the testing of pharmacological treatments for cognition, particularly in patients whose psychosis is well controlled. Because no effective treatment for cognition in schizophrenia is established, animal studies are used to suggest molecular targets, supplemented by clinical data that have indicated molecular mechanisms in schizophrenia. The drugs under investigation are being tested as co-treatments, along with open administration of antipsychotic drugs.
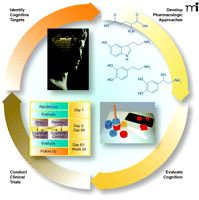
The ongoing search to understand the bases and solutions to schizophrenia. The MATRICS initiative grew out of a series of workgroups sponsored by the National Institutes of Mental Health, attended by academic, industry, and government representatives. The complex disorder, affecting approximately one percent of the population, calls for concerted research from multiple disciplines. See text for details.
The target most favored for cognition is augmentation of central cholinergic function (i.e., nicotinic and/or muscarinic acetylcholine receptors). The highest densities of nicotinic acetylcholine receptors (nAChRs) are in the medial temporal lobe, an area known to be important in schizophrenia pathology; expression of the α7 nicotinic receptor appears to be reduced in the illness. Administration of a partial nicotinic agonist has shown promise in reversing some aspects of cognitive dysfunction (6). Muscarinic acetylcholine receptors (mAChRs) are also compelling targets for cognitive improvement in schizophrenia for a number of reasons: their pharmacology in animals; their efficacy (albeit of small magnitude) in Alzheimer’s disease (AD); the actions of xanomeline in schizophrenia (7); and the cognitive decline that occurs with muscarinic antagonist treatments. Selective muscarinic agonists are being tested for therapeutic effects in schizophrenia.
Drugs that enhance glutamatergic plasticity in brain, especially acting at or affecting the N-methyl-D-aspartate (NMDA) receptor, provide a strong rationale for improving cognition. Augmentation of NMDA signaling can, under certain experimental circumstances, improve cognition in animals. Ketamine, an NMDA antagonist, adversely affects cognition in normal and schizophrenic persons (8). Given the complexity of the glutamatergic synapse, drugs may affect synaptic function by acting at the NMDA receptor itself or at its co-agonist (i.e., glycine) site; by interfering with receptor trafficking; or by engaging downstream signaling via metabotropic glutamate (mGlu) receptors. Candidates for cognitive enhancement acting through the NMDA receptor include the amphakines, D-serine, blockers of glycine reuptake, and drugs that affect mGlu receptors. An example of this latter type, the mGlu2/3 receptor agonist LY2140023 (see below), has already demonstrated antipsychotic activity and a profile favorable for cognitive enhancement (9). There exist additional pharmacological categories of putative cognition enhancers identified in animal behavioral studies; whether any of these drugs will be successful in human translation remains to be seen.
The medical need for drug discovery in schizophrenia is vast. The field has advanced by developing component symptom targets, separately modeled in animals, that are thus believed to be pharmacologically independent. The efficacy of drug candidates will not only have to include neuropsychological improvements of performance, but will also need to encompass improvements in psychosocial function. Pharmacological approaches to cognitive dysfunction in the illness may prove vital in mitigating the overwhelming disease burden of schizophrenia.
Allosteric Activators of Muscarinic Receptors
One of the most exciting developments in recent years in developing novel treatments for schizophrenia comes from clinical studies with agonists of mAChRs. A large multicenter trial of patients suffering from AD yielded the surprising finding that xanomeline, an M1/M4-preferring mAChR agonist, had robust therapeutic effects in reducing psychotic symptoms and behavioral disturbances (10, 11). These effects were dose-dependent, and included reductions in vocal outbursts, suspiciousness, delusions, hallucinations, paranoia, and other behavioral disturbances that share similarities with the positive symptoms observed in schizophrenia. Indeed, a growing body of clinical evidence, primarily focusing on acetylcholin-esterase (AChE) inhibitors, corroborates commonalities among behavioral disturbances and psychotic symptoms in patients suffering from AD as well as Lewy body dementia, Parkinson’s disease dementia, and vascular dementia (12–18).
Subsequent investigation of xanomeline, in a double-blind, placebo-controlled clinical study, established the efficacy of the mAChR agonist in improving both positive and negative symptoms in schizophrenic patients (19). Consistent with these findings, antimuscarinic agents induce psychosis, confusion, and cognitive deficits in humans (20). Together, these studies provide strong clinical validation of mAChR agonists as novel therapeutic agents for treatment of psychosis, cognitive defects, and other behavioral disturbances in a broad range of disorders including schizophrenia, AD, and other neurodegenerative disorders. Unfortunately, xanomeline lacks true M1/M4 specificity, and because it has significant affinity and efficacy at M2, M3, and M5 receptors, it suffers from the same peripheral adverse effects as the AChE inhibitors and traditional mAChR agonists, likely reflecting activation of M2 and M3 receptors. This dose-limiting toxicity has prevented previous mAChR agonists from progressing through full clinical development.
The ACh binding site is highly conserved among the mAChRs, and despite tremendous efforts from scientists in industry and academic settings, no traditional (orthosteric) agonists have emerged that bind with sufficiently high selectivity at the M1 or M4. In recent years, we have focused on a novel approach to develop compounds that are highly selective for activating individual mAChR subtypes. Our goal has been to discover novel compounds that increase receptor activity by acting at allosteric sites of mAChRs, thereby avoiding barriers to ligand specificity posed by the highly conserved ACh binding site per se (Figure 3⇓). This strategy has resulted in highly selective allosteric potentiators of the M4 mAChR (21). These compounds do not activate M4 directly (in the absence of agonist) but dramatically potentiate activation of the receptor by ACh. The most robust potentiation is induced by a compound termed VU10010, which induces a fiftyfold leftward shift of the ACh concentration response curve (Figure 4⇓). Interestingly, VU10010 shows an EC50 near 100 nM for M4, but manifests no activity at any other mAChR subtype, even at concentrations as high as 100 μM. VU10010 has no detectable affinity for the ACh binding site but allosterically increases affinity of ACh. We have now developed a series of compounds that have robust CNS activity and reduce amphetamine-induced hyperlocomotor activity, an animal model predictive of potential antipsychotic activity and responsive to xanomeline. In addition, we have found novel allosteric potentiators of the M1 mAChR. As these compounds are further developed, we plan to directly compare the relative effects of allosteric potentiation of M1 and of M4 across the full range of activities that have been established for the M1/M4 agonist in animal models.
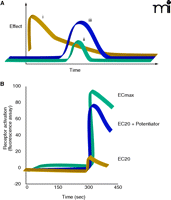
Why bother with allosteric modulation? (A) The response curve for an agonist analog (i) typically reflects pharmacokinetic parameters that fail to replicate the time course of the endogenous agonist (ii). On the other hand, because allosteric modulators, such as agonist potentiators (iii), act by definition to alter the interaction of the endogenous agonist with its receptor per se, the magnitude of the pharmacologically observed effect can be modulated without perturbation of the time course (or localization) of agonism. (B) Hundreds of thousands of compounds have been screened for biological activity at mGlu and mACh receptor subtypes. Allosteric modulators (here, potentiators) have been identified by their ability to raise levels of receptor activation at a given “effective concentration” (EC) of agonist. See text for details.
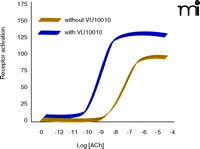
VU10010 acts as an allosteric potentiator of the M4 mACh receptor. VU10010 induces a 50-fold leftward shift of the ACh concentration response curve. VU10010 has an EC50 near 100 nM for the M4 receptor subtype but no activity at any other subtype even at concentrations as high as 100 μM. VU10010 has no detectable affinity for the ACh binding site but allosterically increases affinity of the M4 subtype for ACh. This remarkable specificity, along with the concomitant advantages of allosteric modulation (see Figure 3A), may conform to the ligand characteristics necessary to target pathways and symptoms of complex diseases such as schizophrenia. See text for details.
In addition to allosteric potentiators, we have recently characterized a novel allosteric agonist, termed TBPB, that is highly selective for M1. Unlike the allosteric potentiators, TBPB directly activates the receptor rather than potentiating the effect of ACh. However, like the allosteric potentiators, TBPB acts at a site that is distinct from that of ACh and is highly selective for M1 relative to other mAChR subtypes. Interestingly, TBPB has activity, in brain slices and in vivo, consistent with selective activation of M1 and has robust effects in a range of animal models that are commonly used to predict antipsychotic activity. The development of small-molecule ligands that are highly selective for individual mAChR subtypes is an exciting development, suggesting that selective allosteric activators of M1 and/or M4 may lead to new treatments of schizophrenia.
Metabotropic Glutamate Receptors as Novel Targets
Glutamate receptors are classified into two types. The ionotropic glutamate (iGlu) receptors are ligand-gated ion channels, whereas the metabotropic glutamate (mGlu) receptors are G protein–coupled receptors. The mGlu receptors fall into three subgroups, based on their pharmacology and the molecular structures. Group I mGlu receptors include mGlu1 and mGlu5 (coupled to phosphoinositide hydrolysis); group II mGlu receptors include mGlu2 and mGlu3 (linked negatively to cAMP formation); and group III mGlu receptors include mGlu4, mGlu6, mGlu7, and mGlu8 receptor subtypes (linked negatively to cAMP formation).
In contrast to iGlu receptors, which directly depolarize the post-synaptic cell, mGlu receptors function to indirectly modulate glutamatergic transmission by pre- and post-synaptic, and glial mechanisms (22). These modulatory roles, along with the relative distributions of mGlu receptors (e.g., mGlu2, -3, and -5 receptors are highly expressed in the forebrain), have led to the concept that mGlu receptors are potential targets for therapeutic intervention of brain excitability and plasticity in psychiatric (and neurological) diseases (23, 24).
Substantial progress has been made in the pharmacology and cellular actions of the group II (mGlu2/3) receptors, greatly facilitating appraisal of their potential as therapeutic agents (25, 26). For example, agonists for mGlu2/3 receptors decrease the evoked release of glutamate and indicate a key role for these receptor subtypes in suppressing neuronal excitability, which may be relevant to pathological states that result from excessive release of glutamate (22, 27). Pathological glutamate release, or altered glutamate receptor activity, signaling, and expression, have been implicated in many clinical conditions, including schizophrenia (28).
Phencyclidine-like drugs increase cortical excitability (e.g., as measured using blood flow imaging), which is positively correlated to induction of psychosis (29, 30), and phencyclidine (PCP) and ketamine are open channel blockers of the NMDA receptor complex (31). These observations collectively serve as the basis of a glutamate hypothesis of schizophrenia. Specifically, novel glutamatergic drugs that suppress hyperexcitation in the limbic cortical regions might be antipsychotic in humans (30, 32). As mGlu2/3 receptor agonists suppress glutamatergic excitations in the limbic cortex and have antipsychotic activity in certain animal models, they have been used to explore the role of glutamate modulation in schizophrenia (33). Most recently, promising clinical results against positive and negative symptoms in schizophrenics were reported for a novel mGlu2/3 receptor agonist (34). This report has greatly increased interest in non-dopamine approaches to schizophrenia, as it indicates that glutamatergic approaches to may be clinically viable (28).
The antipsychotic actions of multiple mGlu2/3 receptor agonists have been explored in animal models (35–38), and the cellular mechanisms that may underlie these actions have been previously reported and continue to be elucidated (39). (Table 1⇓ provides a summary.) Notably, mGlu2/3 receptor agonists block the effects of PCP and related drugs (e.g., ketamine) on behavior, including increases in locomotor behavior, cognitive disruptions, and disruptions of attentional behavior [see (33)]. At the cellular level, mGlu2/3 receptor agonists block NMDA antagonist–induced neuronal degeneration and neurotransmitter release in vivo (e.g., glutamate, dopamine, serotonin, and norepinephrine) (35, 40). Actions of other psychostimulant agents, such as amphetamine and the 5HT2A receptor agonist DOI, have also been used to explore antipsychotic activity in animals; mGlu2/3 receptor agonists block certain behavioral actions of these agents as well (33). The ability of mGlu2/3 receptor agonists to reverse behaviors across mechanistically distinct psychostimulants is an interesting feature. In certain brain regions and models, mGlu2/3 receptor agonists also block the in vivo release of various neurotransmitters otherwise released as a consequence of NMDA receptor blockade. The multifaceted role of glutamate in the direct and indirect modulation of many neurotransmitter systems in the brain may signify multiple neuronal mechanisms in mGlu-mediated antipsychotic approaches to treatment. In addition, mGlu2/3 receptor agonists potently suppress glutamate release in rat prefrontal cortex, as reflected by excitatory post-synaptic potentials (EPSPs) induced via 5HT2A receptors (41, 42). These actions of mGlu2/3 receptor agonists are also blocked by a selective mGlu2/3 antagonist (which increases EPSPs per se). Intriguingly, atypical antipsychotic drugs, such as clozapine, also suppress 5HT2A receptor–induced EPSPs in the prefrontal cortex, thus suggesting functional commonality between atypical antipsychotic drugs and mGlu2/3 receptor agonists (32, 33). Recently, 5HT2A receptors and mGlu2 receptors were reported to form functional heterodimers that can be pharmacologically modulated by mGlu2/3 receptor agonists and antagonists (39). In schizophrenic brains, the 5HT2A receptors are upregulated, and mGlu2 receptors are downregulated, suggesting molecular bases for predisposition to psychosis.
Effects of mGlu2/3 Receptor Agonists Relevant to Schizophreniaa
It is not clear which of the immediate or downstream actions of mGlu2/3 receptor activation are most important to therapeutic potential. Given the high variability and complex time course of symptoms in schizophrenia, it will be a challenge to link particular aspects of symptoms to mechanisms of receptor signaling and neurotransmission. There are many important questions left to answer in the area of glutamate modulation alone; future pre-clinical and clinical pharmacological investigations with mGlu2/3 receptor agonists and other promising agents (such as selective mGlu2 and mGlu5 receptor allosteric modulators) will help provide answers.
Therapeutic Potential of Inhibitors of the Glycine Transporter
The N-methyl-D-aspartate (NMDA) receptor antagonist PCP has been shown to induce the positive, negative, and cognitive symptoms of schizophrenia in healthy patients and elicit a resurgence of symptoms in stable schizophrenics (43). In the clinic, the observation that administration of the NMDA receptor co-agonist glycine provides a modest improvement in schizophrenic patients suggests that increasing NMDA receptor activation may provide a therapeutic benefit (44). These observations led to the NMDA receptor hypofunction hypothesis of schizophrenia (45). According to this hypothesis, any agent that can potentiate NMDA receptor currents, either directly (by acting at modulatory sites of the receptor, such as the glycine co-agonist binding site) or indirectly (by activating other receptors, such as the mGlu5 receptor, known to potentiate NMDA receptor function), has the potential to ameliorate the symptoms of schizophrenia (Figure 5⇓) (46).
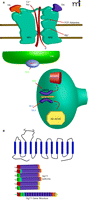
The NMDA receptor hypofunction hypothesis and mechanisms to increase NMDA receptor function. (A) NMDA receptor function may be approached by direct activation (GlyT1 inhibtion) or indirect activation by activating GPCRs (M1 and mGluR5) that physically and functionally interact with the NMDA receptor. The binding site of the co-agonist glycine on the NR1 subunit and the the binding site of the co-agonist glutamate on the NR2 subunit are indicated. (B) The structure of GlyT1, a 12-transmem-brane domain Na+/Cl− dependent neurotransmitter transporter, is schematized along with the gene structure of GlyT1 and the six major iosforms of GlyT1. See text for details.
Glycine is a required co-agonist for the NMDA receptor and modulates NMDA-dependent excitatory neurotransmission; therefore, one approach to enhance NMDA receptor function is to pharmacologically increase synaptic glycine levels (47). In the CNS, synaptic glycine levels are regulated by glycine transporter type (GlyT)-1 and –2, Na+/Cl−-dependent transporters that share 50% homology at the amino acid level (48). Importantly, GlyT1 distribution mirrors NMDA receptor expression, suggesting that GlyT1 is optimally positioned to modulate glycine levels near NMDA receptor–expressing synapses (48).
The modulation of synaptic glycine levels as a therapeutic strategy has strong clinical support. Both glycine and sarcosine, a weak selective GlyT1 inhibitor (Figure 6⇓), have been shown to improve the positive, negative, and cognitive symptoms of schizophrenia; however, poor brain penetration and pharmacokinetics limit their clinical utility (Figure 1⇑) (49, 50). The first GlyT1 inhibitors were analogs of sarcosine, such as NFPS (Figure 6⇓), that provided support for the NMDA receptor hypofunction hypothesis by selectively elevating glycine levels and reversing PCP-induced behaviors in preclinical animal models (43, 44). Nevertheless, a variety of side effects, partly arising from a noncompetitive mode of inhibition, prompted many pharmaceutical companies to launch efforts to identify non-sarcosine-derived GlyT1 inhibitors (51).
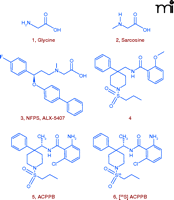
Design of GlyT1 Ligands. Structures of glycine 1 (co-agonist of the NMDA receptor), sarcosine 2 (a weak GlyT1 inhibitor) and NFPS 3 (ALX-5047), the prototypical sarcosine-derived GlyT1 inhibitor, competitive, non-sarcosine-derived GlyT1 inhibitor lead 4, optimized GlyT1 inhibitor 5 (ACPPB), and the radiolabelded congener [35S]ACPPB, 6. See text for details.
To circumvent the problems associated with sarcosine and its analogs, non-sarcosine-derived, competitive GlyT1 inhibitors have been sought through a high-throughput screening campaign, employing a [14C]-glycine uptake scintillation proximity assay (52, 53). A novel compound, from a series of [4-phenyl-1-(propylsulfonyl)piperidin-4-yl]methyl benzamides (Figure 6⇑), was identified as a potent, reversible inhibitor of GlyT1 (IC50 = 135 nM) with high selectivity against GlyT2. An iterative analog library synthesis approach rapidly developed structure-function relationships that led directly to the discovery of ACPPB, a potent (IC50 = 2.1 nM), competitive, and centrally active inhibitor with complete selectivity for GlyT1 from among a battery of receptors, channels, and enzymes. ACPPB selectively increased prefronto-cortical extracellular glycine levels (340% of basal levels) with no effect on other amino acids. By selective blockade of GlyT1, ACPPB significantly enhanced prepulse inhibition of startle in a rodent behavioral model sensitive to antipsychotic treatment (53). These data suggest toxicity issues observed with NFPS and related non-sarcosine-derived congeners related to chemotype and the associated non-competitive mode of inhibition, rather than a fundamental reflection of GlyT1 inhibition.
To further study the detailed pharmacology of these competitive, non-sarcosine based GlyT1 inhibitors and the distribution of GlyT1 in the brain, we prepared an [35S] ACPPB radioligand (54). In this way, ACPPB binding sites in rat and rhesus brain have been revealed and in vivo occupancy assays have been developed for competitive GlyT1 inhibitors. Studies with the radiolabeled ACPPB have also demonstrated the feasibility of developing a PET tracer for GlyT1. These data provide additional strong support for the NMDA receptor hypofunction hypothesis of schizophrenia and the development of novel antipsychotics.
Footnotes
-
↵1 This article is based on a symposium organized for the annual meeting of ASPET at Experiental Biology 2008, San Diego, CA, April 5–9, 2008.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Craig W. Lindsley, PhD, is Associate Professor of Pharmacology & Chemistry and the Director of Medicinal Chemistry, Vanderbilt Program in Drug Discovery. Prior to joining Vanderbilt, Dr. Lindsley established and led the Technology Enabled Synthesis Group within the Medicinal Chemistry Department at Merck, West Point. Research within the Lindsley lab, both at Merck and at Vanderbilt, has focused on the design of allosteric ligands to modulate GPCRs, kinases and lipases with special emphasis on CNS disorders and cancer. Dr. Lindsley has pioneered the development of mGluR5 positive allosteric modulators, allosteric Akt inhibitors and nonsarcosine-derived, competitive GlyT1 inhibitors.

Darryle D. Schoepp, PhD, is Senior Vice President and Franchise Head for Neuroscience at Merck Research Laboratories, with overall responsibility for scientific direction across drug discovery and development in Psychiatric Disorders, Neurology, and Ophthalmology. His research career has focused on glutamate receptor pharmacology, where he has led efforts to discover and investigate novel agents for schizophrenia and other CNS applications.

Carol A. Tamminga, MD, is Professor of Psychiatry, Vice Chair of Clinical Research, and Chief of Translational Neuroscience Research in Schizophrenia at the University of Texas Southwestern Medical School. She holds the Communities Foundation of Texas Chair in Brain Science and directs clinical and preclinical research in schizophrenia. Her clinical research laboratory utilizes a human postmortem brain collection and in vivo human brain imaging. She has been the recipient of numerous grants and has served national granting bodies in multiple capacities.

P. Jeffrey Conn, PhD, is Lee E. Limbird Professor of Pharmacology and founding Director of Vanderbilt Program in Drug Discovery at Vanderbilt Medical Center. During his early academic career he established himself as a leader in studies of neurotransmitter receptors and their roles in regulating brain function in circuits involved in psychiatric and neurological disorders. He has also directed industrial initiatives with a primary mission of facilitating translation of advances in basic science into novel therapeutics. He is Editor in Chief of Molecular Pharmacology, Regional Editor (North America) of Current Neuropharmacology, and serves on the editorial boards of several other international journals. His numerous awards include the NARSAD–Essel Distinguished Investigator Award, the Pharmacia-ASPET Award in Experimental Therapeutics, the ASPET Astellas Award in Translational Pharmacology, and he is among the ISI Most-Cited Scientists in Pharmacology and Toxicology. Through many leadership roles in academia and within the research and drug discovery communities, Dr Conn has greatly strengthened the discipline of pharmacology. His current research is focused on development of novel treatment strategies for schizophrenia, Parkinson’s disease, and other brain disorders. E-mail jeff.conn{at}vanderbilt.edu; fax 615-936-6833.

