The Only Way Is Up: Preventing Opioid Tolerance by Promoting Cell Surface Expression of MOR-DOR Heterodimers?
Our understanding of G protein–coupled receptor (GPCR) dimerization has steadily progressed over the last two decades, revealing new and intriguing insights into receptor dimer biogenesis, trafficking, and function. Dimerization occurs in all three major classes of GPCRs, and in several cases has been reported to be essential for proper function of the receptors in the dimer complex. For example, among the taste receptors, T1R3 must dimerize with T1R2 or T1R1 to produce a functional receptor complex that recognizes sweet (1) or umami (2), respectively. In other cases, receptors trapped inside the Golgi compartment or endoplasmic reticulum can be rescued and transported to the cell surface via dimerization with another receptor (3).
In 2004, Saito and colleagues discovered two receptor families, which they named RTP (receptor transporting protein) and REEP (receptor expression enhancing protein), that induced the functional expression of odorant receptors that are otherwise poorly expressed at the cell surface (4). Shortly thereafter, other members of this family were reported to assist in the functional expression of bitter taste receptor T2R16 (5). More recently, Decaillot et al. have shown that RTP4 acts as a chaperone to target a heterodimer between the μ-opioid and δ-opioid receptor (MOR and DOR, respectively) to the cell surface (6).
Opioid receptors are known to form both homo- and het-erodimers (7). Opioid receptor heterodimers are particularly interesting, because they may provide distinct targets for the development of new opioid drugs. For example, 6′-guanidinonaltrindole (6′-GNTI) is an antagonist at DOR homomers (i.e., either an active monomer or multimeric complex of monomers) but functions as an agonist at the heterodimer of the DOR and κ-opioid receptor (KOR), inducing analgesia in spinal cord (8). Indeed, not only can opioid heterodimers bind ligands with altered affinity and efficacy, they can also activate different G proteins and signaling pathways (9). These observations surrounding opioid receptor heterodimers are quite compelling and could possibly shed light on the molecular mechanisms underlying the major side effects of opioid use, including tolerance and dependence.
Several mechanisms have been implicated in the development of morphine tolerance, including protein kinase C, N-methyl-d-aspartate (NMDA) receptors, and cholecystokinin receptors (10). In addition, within the opioid receptor family, factors implicated in the development of tolerance include the trafficking of MOR and the role of DOR as a “pro-nociceptive” receptor (Figure 1⇓). Several reports have observed that heterodimerization of GPCRs can change the endocytic fate of individual receptors (11, 12). This may be important in light of observations that the failure of the MOR to undergo endocytosis in response to activation by morphine contributes to tolerance and dependence to the drug (13). Another interesting finding has been that antagonists of the DOR receptor, as well as disruption of the DOR gene (14, 15), are able to attenuate the development of morphine tolerance (16). These data suggest that, after the development of morphine tolerance, DORs are pro-nociceptive, even though in the naïve state DOR agonists can have anti-nociceptive properties in certain paradigms (17, 18). This switch from anti- to pro-nociceptive is, perhaps, a receptor trafficking phenomenon as well. In particular, in the opi-oid naïve state, DORs are predominantly expressed in intracellular compartments in neurons that are important for pain transmission. Chronic morphine treatment, however, promotes a redistribution of DORs to the cell surface (19, 20). This redistribution appears to require the MOR, as disruption of the MOR gene (21) or blockade of MOR with d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Phe-Thr-NH2 (CTOP) (19) prevents increased cell-surface expression of DOR. In all, these reports suggest that a functional MOR-DOR heterodimer may contribute in some way to analgesia during the development of tolerance. This is why the recent paper describing how RTP4 improves cell surface targeting of MOR-DOR heterodimers is of special interest.
Decaillot and co-workers have also found that RTP4 decreased the ubiquitination of the MOR-DOR heterodimer (6), which likely effects receptor trafficking by decreasing degradation of the receptor complex. Indeed, DORs are targeted for degradation by at least two distinct mechanisms (Figure 1⇓). First, during the process of synthesis and transport, DORs that are not targeted to cell surface are deglycosylated and ubiquitinated before being targeted for proteosomal degradation (22). Second DORs that reach the surface and then are endocytosed are targeted for degradation in lysosomes via interaction with G protein-coupled receptor–associated sorting protein (GASP) (23), which does not require the receptors to be ubiquitinated (24). In contrast, MORs have the ability to recycle and to become resensitized following endocytosis (23). Although the secretory and postendoyctic fate of the MOR-DOR heterodimer remains unknown, one might hypothesize that RTP4 decreases ubiquitination of the heterodimer and thereby increases its surface expression. Alternatively, as elaborated in a separate viewpoint article by Simmons in this issue of Molecular Interventions (25), Zheng et al. have found that opioid ligand functional selectivity is dependent on the location of the receptor and G proteins in raft or non-raft membrane domains (26). Hence, the RTP-chaperoned MOR-DOR heterodimers might be targeted to different microdomains than are the non-chaperoned homomers, and thus have altered function or trafficking. From this point of view, it will be interesting to investigate the G-protein preference and post-endocytic fate of the RTP-chaperoned MOR-DOR heterodimer, as well as the possibility that RTP and Gα proteins may themselves interact, because MOR-DOR het-erodimers are known to interact with Gα proteins in the ER (27).
Perhaps most intriguingly, it will be interesting to determine the role of the MOR-DOR heterodi-mer in morphine tolerance—especially in light of both the anti-nocicep-tive and pro-nociceptive actions of DOR agonists. Tolerance can occur by several mechanisms, none of which are mutually exclusive. Tolerance could be influenced by degradation of target receptors. Alternatively, tolerance could be manifested by a loss of functional receptors in the absence of degradation (either because receptors are trapped in vesicles, or are on the surface but desensitized or in a microdomain that impedes G-protein coupling). In addition, tolerance can be caused by adaptive changes in downstream signaling pathways that do not change receptor number or receptor coupling but alter the ability of the cell to respond to ligand nonetheless. These adaptive changes, including cAMP superactivation (i.e., enhanced cAMP production and ensuing second-messenger activity), appear to be modulated by trafficking of the receptor. Thus, one can imagine several scenarios in which an increased number of RTP-chaperoned MOR-DOR heterodimers would influence morphine analgesia and tolerance. First, if DOR homomers are pro-nociceptive, but MOR-DOR heterodimers are antinociceptive, then increasing the number of MOR-DOR heterodimers could be beneficial at decreasing tolerance (Figure 2A⇓). Second, if the MOR-DOR heterodimer is the pro-nociceptive DOR that enhances tolerance and dependence, then decreasing the function of RTP4 at this dimer could decrease tolerance (Figure 2B⇓). Third, there could also be a specific trafficking effect due to MOR-DOR heterodimerization. MOR homomers are poorly endocytosed in response to morphine and, thereby, promote adaptive changes in signal transduction, such as cAMP superactivation. If MOR-DOR heterodimers, rather than MOR homomers, are better at endocytosing in response to morphine, then their increased expression could help prevent tolerance by preventing these changes (Figure 2C⇓). Fourth, if the MOR-DOR heterodimer is predominantly recycled rather than targeted for degradation like the DOR, this could further enhance any effects described in three scenarios above.
An additional important question is whether RTP4 could, in some way, be a target for pharmacological intervention with the goal of decreasing opioid tolerance. The observation that members of the RTP and REEP family are expressed ubiquitously (5), would suggest that they act as chaperones for many GPCRs––both homomers and heterodimers. There does appear to be some preference among GPCRs for specific RTPs or REEPs. For example, expression of the MOR-DOR heterodimer is enhanced by RTP4 but not RTP2 (6), whereas T2R16 expression is enhanced by RTP3 and RTP4 but not by REEP1 or REEP3 (5). The protein families of RTPs (4) and REEPs (6), however, do not have many members nor are their sequences diverse (4). Therefore, if more receptors are found to interact with these proteins, lack of selectivity may hinder the ability of these proteins to be viable drug targets. Moreover, more studies need to be performed to determine how RTPs and REEPs exert their function on receptor trafficking. FACS analysis has revealed that expression of REEPs not only increases the number of α2C-adrenergic receptors on the cell-surface but also increases the total number of intact receptors to a similar degree (28). In short, the ratio of intracellular receptors versus surface-expressed receptors does not change with REEP expression, suggesting REEPs do not actually promote increased trafficking but merely increase the total number of receptors. Nevertheless, the research performed by Decaillot and co-workers illustrates once again that heterodimers should be perceived as novel and perhaps tissue selective (and therefore less prone to side effects) drug targets.
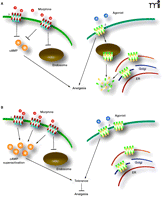
Schematic representation MOR- and DOR-mediated analgesia and tolerance. A. In the naïve state MOR agonists, such as morphine, inhibit cAMP formation and promote analgesia. Agonist activation of surface expressed DORs produces analgesia but many DORs are expressed intracellularly and are targeted for degradation. B. Prolonged activation of MORs causes cAMP superactivation, an early indicator of tolerance development, possibly as a consequence of the failure of MORs to undergo endocytosis and recycle. Chronic morphine also causes an upregulation of surface expressed DORs. At least some of these DORs are pro-nociceptive and contribute to morphine tolerance.
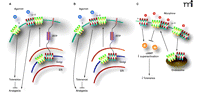
Schematic representation of how surface expressed MOR-DOR heterodimers could influence morphine tolerance. DORs forming heterodim-ers with MOR are chaperoned to the cell surface by interaction with RTP. It is conceivable that (A) the DOR-MOR heterodimers are anti-nociceptive and the DORs are pro-nociceptive, or (B) that DORs are anti-nociceptive and DOR-MORs are pro-nociceptive, thus contributing to tolerance. C. Heterodimers between MOR-DOR may have alternate signaling and/or trafficking properties that differ from those of MOR homomers, possibly reducing the effects of cAMP superacti-vation and, thereby, reducing tolerance.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Jennifer L. Whistler, PhD, is a Principal Investigator at the Ernest Gallo Clinic and Research Center and an Associate Professor of Neurology at the University of California San Francisco. Her research interests center on the signaling and trafficking properties of GPCRs important in addiction and other neuropsychiatric diseases. Address comments to Jwhistler{at}gallo.ucsf.edu; fax 510-985-3101.

Richard M. van Rijn, PhD, is a Post-doctoral research scientist in the laboratory of Jennifer Whistler at the Ernest Gallo Clinic and Research Center. His research interests include the oligomerization of G protein–coupled receptors.

