Orthosteric/Allosteric Bitopic Ligands
Going Hybrid at GPCRs
Abstract
G protein–coupled receptors (GPCRs) can adopt multiple biologically active states that can be differentially stabilized by ligands that bind to topographically distinct sites (e.g., orthosteric ligands and allosteric modulators). Recent studies in the field are now demonstrating the utility of linking orthosteric and allosteric pharmacophores to yield hybrid, or bitopic, ligands, with improved affinity and/or receptor subtype selectivity. Interestingly, this approach can also engender functional selectivity in the actions of orthosteric ligands, highlighting a viable means of further sculpting GPCR ligand responses. Indeed, some previously identified functionally selective agonists may actually represent hitherto unappreciated bitopic ligands.
Introduction
G protein–coupled receptors (GPCRs) constitute the largest family of cell surface receptors in the human genome (1). Owing to their ubiquitous cell surface distribution and their contributions to multiple physiological and pathophysiological processes, GPCRs have frequently been exploited as drug targets, currently accounting for around thirty percent of all medicines on the market (2). Even so, only around one-fifth of the total complement of GPCRs in the genome have been established as therapeutic targets, thus indicating a substantial potential for further drug discovery amid this important family of receptors.
Structurally, all GPCRs share a characteristic architecture, including seven transmembrane-spanning domains, which has evolved to accommodate the dual roles of extracellular ligand recognition and intracellular signal transduction. Despite this common architecture, however, GPCRs display enormous functional versatility, responding to light, ions, odorant molecules, biogenic amines, lipids, nucleotides, peptides, and large proteins. Moreover, the list of membrane-delimited and intracellular coupling partners is constantly expanding, highlighting the remarkable conformational flexibility inherent in GPCRs. Their participation in a vast array of protein–ligand and protein–protein interactions indicates that GPCRs must be able to present discrete sites, assume multiple conformations, and by extension, offer the potential for multiple modes of drug targeting. By and large, however, the traditional approach to GPCR-based drug discovery has focused on orthosteric sites, that is, the binding sites recognized by endogenous agonists. Admittedly, the chemical structure of an endogenous orthosteric ligand may be the most logical starting template for the design of drugs that may act as agonists or antagonists, but it is imperative to understand that such a design strategy cannot always address issues of selectivity, given that the orthosteric site is likely to be highly conserved across receptor subtypes.
The last two decades have witnessed an explosion in our understanding of new paradigms of GPCR behaviors. For instance, it is now well established that GPCRs can display constitutive (i.e., ligand-independent) activity, which can be differentially modulated by alternative types of ligands (3). GPCRs can also possess topographically distinct allosteric sites, recognized by structurally diverse allosteric modulators, which regulate the affinity and/or efficacy of co-bound orthosteric ligands (4, 5). New insights into the intermolecular associability of GPCRs, with one another and with cellular accessory proteins, imply an unexpectedly broad spectrum of complexes that may presage the potential for orthosteric and allosteric ligands to attain signal pathway selectivity as a basis for new therapeutics (6, 7). These new paradigms have spurred a dramatic reevaluation of approaches to design and develop selective GPCR ligands. Here, we will address some of these issues and explore recent efforts to exploit multiple modes of ligand–receptor interactions in the design of small-molecule modulators of GPCR function.
Messages and Addresses: Bivalent GPCR Ligands
Many of the classic approaches to selective GPCR-based drug design have entailed structure-activity studies and can generally be described as variations of the “message-address” concept (8). Although originally coined to describe the action of peptide hormones at their cognate receptors, the more general principle is that a “message” component of the ligand molecule exists to effect signal transduction, whereas an “address” component, located adjacent to the message, provides additional binding affinity. Most relevant to the current discussion is the nature of the regions on the receptor that engage the “message and address” components. More specifically, whereas the message component of a given ligand may be presumed to interact primarily at the receptor’s (highly conserved) orthosteric site, the address component may be able to engage receptor sequences that have evolutionarily diverged across receptor subtypes. By invoking these two considerations, ligand design efforts can incorporate selectivity as well as binding affinity as design criteria. Indeed, the consideration of the distinct message and address components of ligands, along with their respective interactions at receptor sites, has inspired design efforts in which distinct pharmacophores are chemically linked (9). The synthesis of bivalent ligands gained particular prominence, beginning in the 1980s, as a means of developing highly selective compounds for GPCRs (10), although the approach dates back to the first half of the last century with studies on ligand-gated ion channels (11). The concept was most notably extended to GPCRs by Portoghese and colleagues and their studies of opioid receptors [see (12–14)], but it has also been applied to others, including muscarinic receptors (15–17), serotonin receptors (18, 19) and GnRH receptors (20). Figure 1⇓ illustrates some examples of compounds synthesized on the basis of a bivalent ligand approach.
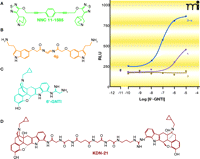
Some examples of bivalent orthosteric GPCR ligands. Structures are shown for (A) a homobivalent muscarinic receptor agonist; (B) a homobivalent serotonin receptor agonist; (C) a δ/κ opioid heterodimer–selective agonist; and (D) a heterobivalent dimer-selective antagonist. The inset shows that 6′-GNTI has higher efficacy for promoting intracellular calcium mobilization in HEK cells containing the δ/κ heterodimer over cells individually expressing either the δ or κ opioid receptor. Data replotted from (22).
Homobivalent Ligands
If the bivalent ligand is constructed from two pharmacophores that are identical (i.e., a “message-message” paradigm) the effective concentration of the active pharmacophore within the immediate region of the receptor will be elevated, because each of the two pharmacophores will raise the probability of a productive binding event. If the homobivalent construction allows both pharmacophores concomitantly to engage adjacent receptor sites, one can anticipate a dramatic increase in binding affinity, in theory equal to the product of the binding contributions of the individual pharmacophores (9). In this way, the potential of multivalent ligand design resonates intriguingly with mounting evidence that many GPCRs may exist as dimers and higher-order oligomers.
Heterobivalent Ligands
A growing body of data supporting heterodimeric GPCR complexes has spurred further studies into heterobivalent ligands, as illustrated by two striking examples presented recently by Porthoghese and colleagues (Figure 1⇑). The first of these concerns 6′-guanidinonaltrindole (6′-GNTI), which was originally designed to be a κ opioid receptor agonist on the basis of the message-address concept: the guanidinium group was envisaged as the address component of the ligand, whereas the naltrindole represents the pharmacophore message (21). Interestingly, subsequent studies revealed 6′-GNTI to be a more potent and selective agonist of the δ–κ opioid receptor heterodimer (22). A second example of a GPCR heterodimer–selective ligand is KDN-21 (Figure 1⇑), a heterobivalent ligand designed to contain both κ-selective and δ-selective pharmacophores, separated by a linker (23).
The synthesis of other heterobivalent ligands has been predicated on a desire to combine pharmacophores that result in a common therapeutic endpoint, or simply as novel biological probes of receptor function. For instance, the conjugation of the β2 adrenergic receptor agonist isoproterenol to the A1 adenosine receptor agonist adenosine has been attempted in an effort to derive novel tools for modulating cardiac arrhythmias (24). The joining of antagonists for the V1a vasopressin receptor and the B2a bradykinin receptor has also been described (25). In another example, which combines both agonist and antagonist pharmacophores (in this case, serotonin and 1-naphthylpiperazine, respectively) into a single bivalent ligand, the 5-HT1B/1D serotonin receptor subtype was selectively targeted (26). Such studies have established that bivalent ligands, constructed with appropriate pharmacophores and linker, can provide significant gains in selectivity and/or potency relative to simpler pharmacophores. But again, as discussed above, research has predominantly focused on the orthosteric nature of the chosen pharmacophores, with less emphasis placed on the “accessory” sites through which these ligands may interact and manifest selectivity. Similarly, little attention has been given to the existence of unique GPCR conformational states that may contribute to the biological activity of bivalent ligands. As a prelude to discussing this notion, it is necessary to review briefly the phenomena of allostery and functional selectivity at GPCRs.
Novel Sites and Novel States: Allosteric Modulation and Functional Selectivity
With the advent of functional high-throughput screening in recent years, there has been a tremendous increase in the identification of small molecules that target allosteric sites on GPCRs (27) and thereby modulate orthosteric binding as well as interactions with regulatory proteins (e.g., G proteins, arrestins, and other GPCRs). Depending on the nature of conformational changes engendered by allosteric modulators, the affinity and/or efficacy of orthosteric ligands can be altered (5), and this alteration may be determined by multiple allosteric interactions, each of which may contribute in its way (e.g., positively or negatively) to the conformation and functionality of the given receptor (28). Even in the absence of orthosteric ligand, some allosteric modulators can themselves act as agonists or inverse agonists (29), highlighting the multiplicity of binding modalities that can control receptor function. For example, the novel small molecule LY2033298 is not only a positive and selective modulator of acetylcholine agonism (30), but also an allosteric agonist, in the absence of acetylcholine, of the M4 muscarinic acetylcholine receptor (31). This latter finding indicates that it is possible to find multiple properties (e.g., affinity modulation, efficacy modulation, allosteric agonism, and inverse agonism) encoded in the same allosteric ligand.
In order to describe allosteric effects, various mass-action models have been proposed, set around a ternary complex that includes two ligands (orthosteric and allosteric) and a receptor that adopts one or more conformational states (32–34). In addition to the affinities of both ligands for their respective binding sites, these models incorporate one or more “cooperativity factors” to account for the effect(s) that each of the two ligand brings to bear upon the interaction of the other with the receptor; these cooperativity factors thus allow for mechanistic complexity beyond competitive reaction schemes. This has significant practical implications for the design of novel modulators, as both modulator affinity and cooperativity need to be considered in structure-activity studies. A particular challenge in this regard, unique to allosteric interactions, is the issue of “probe dependence,” whereby the allosteric interaction can change depending on the type of orthosteric ligand that is used to probe receptor function (35, 36). For instance, the musinteraction between LY2033298 and acetylcholine at the M4 carinic receptor is characterized by positive cooperativity, whereas LY2033298 manifests neutral cooperativity with respect to receptor binding of the antagonist quinuclidinyl benzilate (31). Thus, if only the latter orthosteric ligand (i.e., quinuclidinyl benzilate) had been used for screening, the activity of LY2033298 as an allosteric modulator would have gone undetected.
Despite the conceptual challenges they pose for ligand designers, allosteric modulators offer numerous potential advantages that will hopefully validate the increased emphasis they are receiving. The most obvious advantage is that allosteric modulators can achieve greater receptor subtype selectivity—whether in terms of binding or cooperative effects—than can orthosteric ligands. If the modulator does not possess any agonistic properties, it will remain quiescent in the absence of endogenous orthosteric tone and will thus function solely within the spatial and temporal properties of endogenous physiological signaling. If the modulator is characterized by limited positive or negative cooperativity, its pharmacological effect will also be limited even in the presence of high doses, with a lower likelihood towards target-based toxicity compared to orthosteric ligands.
Concomitant with increasing interest in GPCR allosteric sites as novel drug targets has been the identification of ligand–receptor interactions that deviate, in terms of some observable receptor function, from classic receptor theory. Many instances of such deviations may reflect a flaw in our traditional, theoretical positing of a single active receptor state. For example, propranolol, which was discovered over forty years ago as a β-adrenergic receptor “blocker” has more recently been found to be an inverse agonist of G protein–dependent pathways and a positive agonist of β-arrestin–mediated ERK1/2 phosphorylation (37, 38). This striking reversal in our understanding of propranolol efficacy belies the traditional description of the GPCR as a simple on-off switch. Instead, the receptor appears to adopt a multitude of states, each linked to distinct functional outcomes. The preferential stabilization of a given subset of these states will thus be determined by the particular constellation of bound ligands. Many examples now exist to support the concept of a single ligand that promotes distinct roles for a given receptor, depending on the pathway in which the receptor is engaged (39). Even within a given signaling pathway a receptor may manifest distinctive roles, depending upon whether the pathway is triggered constitutively in the absence of ligand, relative to the presence of ligand (40). The ability of different ligands to engender discrete cell signaling activities at a given GPCR has been termed “stimulus-trafficking”, “biased agonism,” or “functional selectivity” (7). A key diagnostic indicator of functional selectivity is the reversal of ranked agonist potencies or maximal effects at a given receptor examined across alternative signaling pathways. The variability of agonist rankings (e.g., in terms of potency or maximal effect) upon the specified context of cell signaling highlights the need for functional screening of drug candidates under multiple conditions. In other words, any GPCR as drug target must be considered in terms of the particular active state that it adopts in the service of the given particular signaling pathway. Our growing understanding of functional selectivity contradicts the traditional concept of a GPCR as a singularly defined “molecular target.”
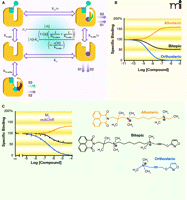
Figure 2⇓ summarizes some of the key characteristics of allosteric modulation and functional selectivity. It is important to note, however, that both phenomena fundamentally reflect a common mechanism, namely, a conformational change in a GPCR, evoked by one ligand, that affects the interactive properties of the receptor with a second ligand. In the case of classic allosteric modulation, the first ligand is an allosteric modulator, and the second is the orthosteric ligand; in the case of functional selectivity, the first ligand could be an agonist, and the second “ligand” might be an effector that mediates a particular signaling pathway. It follows that allosteric modulators can themselves engender functional selectivity, either in the absence or (perhaps more interestingly) in the presence of orthosteric ligand (36). Given the growing number of allosteric modulators known for GPCRs, we may discover novel agents for selectively targeting discrete signaling states.

Characteristics of GPCR allosteric modulation and functional selectivity. See text for details.
“Hybrid Technology”: Bitopic GPCR Ligands
The most obvious application of allosteric ligands is to modulate endogenous agonism, assuming there is sufficient endogenous orthosteric tone to be modulated. In terms of therapeutic goals, orthosteric ligand and modulator are best considered in concert. Despite caveats with respect to probe dependence as outlined above, the phenomenon of allosteric modulation suggests that it is theoretically possible to engender selectivity in the actions of an otherwise non-selective orthosteric ligand. Rather than resorting to the co-administration of two separate bioactive molecules, however, we may opt to encode both orthosteric and allosteric recognition properties within a single therapeutic agent, producing a bitopic molecule that bridges two topographically distinct ligand-binding domains. Although this is similar to the bivalent ligand approach, the term “bitopic” is used to explicitly acknowledge that recognition of an allosteric site is one of the desirable properties of the hybrid molecule. Given that this concept is quite recent, reports of the phenomenon to date have been restricted to studies of the M2 muscarinic acetylcholine receptor (41–44), a Family A GPCR for which there is substantial pharmacological and mutagenesis data supporting the presence of at least one “prototypical” small-molecule allosteric binding pocket (45).
A pioneering example of the empirical design of a bitopic ligand comes from the work of De Amici, Holzgrabe, and colleagues (42), who synthesized hybrids comprising moieties based on oxotremorine (i.e., an orthosteric muscarinic agonist) coupled to hexamethonium-derived allosteric modulators (i.e., ligands of the aforementioned “prototypical” allosteric binding site on the M2 muscarinic receptor). The resulting hybrid molecules display both allosteric and orthosteric binding properties, although the signaling efficacy of the orthosteric moiety can appear to be substantially reduced relative to the parent agonist. In a subsequent study, the first formal description of a bitopic mechanism was presented (Figure 3A⇓) (5). In this model, two equilibrium dissociation constants are invoked to describe engagement of the receptor by the bitopic ligand. In the first instance, that of the bitopic binding mode, the dissociation constant (KB-ortho) is formally analogous to a description of orthosteric binding in the sense that steric hindrance prevents the binding of other orthosteric ligands. In the second case (KB-allo), the bitopic compound functions solely as a ligand at the allosteric binding site, so that the orthosteric pocket is still available for binding of any orthosteric ligand (e.g., the endogenous agonist); for this instance, the model must include the constant α, a cooperativity factor that quantifies the magnitude and direction of the allosteric effect of the bitopic ligand upon orthosteric ligand binding. Values of α > 1 denote positive cooperativity, whereas negative cooperativity is defined by 0 < α < 1, and α = 1 denotes neutral cooperativity. At the level of receptor occupancy, therefore, the bitopic model is a composite of the standard competitive model (governed by KA and KB-ortho) and the classical allosteric model (governed by KA, KB-allo, and α). The extent to which one behavior predominates over the other is ultimately determined by the values of the relevant model parameters (5). Figure 3B⇓ shows a theoretical example of conjugating a positive allosteric modulator to an orthosteric ligand. Binding isotherms, generated for a fixed concentration of an orthosteric probe, predict that the effect of the bitopic ligand in this particular instance would be intermediate between that of a positive modulator and that of a simple competitive orthosteric inhibitor. Importantly, a recent proof-of-concept study by Antony and colleagues (41), in which different types of allosteric modulators were conjugated to iperoxo, an orthosteric muscarinic receptor agonist, has confirmed the validity of the bitopic model. In particular, the hybrid ligands produced in that study conformed to predicted profiles of bitopic binding behavior when tested against an orthosteric probe [the antagonist N-methylscopolamine (NMS)] (Figure 3C⇓).
A bitopic, orthosteric/allosteric, model of GPCR ligand action predicts novel pharmacological properties. (A) A bitopic scheme and associated equation describing fractional equilibrium occupancy (ρA) of orthosteric probe (circle) in the presence of bitopic ligand [square (allosteric pharmacophore) linked to circle (orthosteric pharmacophore)]. The curved arrows denote the potential for allosteric modulation. Also shown is the potential for differential signaling (S) between an orthosteric and a bitopic ligand. (B) Simulations based on the bitopic model with the following parameters: [A] = KA =1 nM; KB-ortho = 3 nM; KB-allo = 30 nM; α = 4. (C) Experimental proof of concept of a bitopic binding phenomenon, as determined at the M2 muscarinic acetylcholine receptor. The indicated ligands were each tested against the equilibrium binding of the orthosteric probe NMS. Data replotted from (41).
As with traditional bivalent ligands, it may be expected that bitopic ligands will display substantially increased affinity for their target receptor compared to the binding affinities of the relevant individual pharmacophores. This is indeed the case with the bitopic ligand THRX-160209 (Figure 4A⇓), which has a binding affinity for the M2 muscarinic receptor that is several orders of magnitude higher than either its orthosteric (3-benzhydryl pyrrolidine; 3-BHP) or allosteric (4-aminobenzylpiperidine; 4-ABP) moieties (43). However, such an expected gain in binding affinity was not seen in the study by Antony and coworkers (41), where two different bitopic ligands displayed affinities similar to those of the corresponding monovalent allosteric pharmacophores and lower than that of the orthosteric pharmacophore, iperoxo (Figure 4B⇓). The differences noted between the two studies may relate to differences in the nature of the orthosteric and allosteric pharmacophores that were combined to yield the bitopic ligands. Specifically, the orthosteric moiety (i.e., 3-BHP) within THRX-160209 is an antagonist, and the allosteric moiety (i.e., 4-ABP) is a negative allosteric modulator; both pharmacophores would thus preferentially interact with a similar, inactive, state of the M2 muscarinic receptor. In contrast, the hybrid molecules reported in the study of Antony and colleagues (41) comprised an orthosteric agonist (iperoxo) conjugated to a negative allosteric modulator (with respect to agonist ligands); in this case, the individual pharmacophores are likely to prefer different states of the receptor, thus incurring a thermodynamic cost with respect to the resulting affinity of the bitopic ligand.
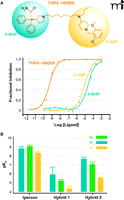
Bitopic ligand design as a means for optimized affinity or selectivity. (A) Effects of the bitopic ligand THRX-160209 and its constituent orthosteric (3-BHP) or allosteric (4-ABP) moieties on the binding of the orthosteric probe NMS at the M2 muscarinic receptor. Adapted from from (43). (B) Loss of affinity, but gain in selectivity, as exemplified by the bitopic ligands, “hybrid 1” (2-{3-[1-(6-{1,1-Dimethyl-1-[4-(isoxazol-3-yloxy)but-2-ynyl]-ammonium}hexyl)-1,1 dimethylammonio]propyl}isoindoline-1,3-dione dibromide) and “hybrid 2” (2-{3-[1-(6-{1,1-Dimethyl-1-[4-(isoxazol-3-yloxy)but-2-ynyl]-ammonium}hexyl)-1,1-dimethylammonio]-2,2-dimethylpropyl}-benzo[de] isoquinoline-1,3-dione dibromide), relative to the non-selective orthosteric agonist iperoxo. Native muscarinic acetylcholine receptors are expressed in guinea pig left atrium (M2), rabbit vas deferens (M1), and guinea pig ileum (M3). Adapted from (41).
Despite the possibility of variable effects in ligand affinity, the important advantage in pursuing the development of bitopic ligands is the degree of selectivity that can be gained by exploiting both orthosteric and allosteric modalities of binding. As shown in Figure 4B⇑, despite an overall loss in binding affinity, bitopic molecules have been developed that manifest substantially increased selectivity for native M2 muscarinic receptors over M3 receptors (increases by at least one to two orders of magnitude), and a slight preference over M1 receptors as well (41). Intriguingly, studies of M2 muscarinic receptor–mediated whole cell dynamic mass redistribution suggest that bitopic ligands may activate only a subset of the intracellular pathways activated by the parent (i.e., unconjugated) orthosteric agonist (e.g., acetylcholine or iperoxo) (41). This intriguing interpretation awaits further mechanistic investigation.
Bitopic Ligands Rediscovered?
The possibility of engendering functional selectivity in the actions of an orthosteric ligand by coupling it to an allosteric modulator raises the intriguing question of whether previously reported functionally selective agonists actually represent hitherto unappreciated bitopic ligands. A recent study, again utilizing the M2 muscarinic receptor as a model system, suggests that this may indeed be the case. McN-A-343 (Figure 5⇓) is one of the earliest known examples of a muscarinic receptor agonist that displays substantial differences in efficacy across receptor subtypes; for instance, it is often reported to be a full agonist in assays of M1 or M4 receptor signaling, but a weak partial agonist at the M2 receptor (46). Although its interactive properties with orthosteric agonists and antagonists are compatible with a competitive mechanism over a wide range of concentrations, the compound can also allosterically modulate the M2 receptor, at least at high concentrations, when access to the orthosteric site is precluded in the presence of the antagonist NMS (47–49). Moreover, a recent study has suggested that McN-A-343 can traffic stimuli preferentially to Gα15 proteins, relative to Gαi proteins, when compared to classic mAChR agonists (50). Attempts to clarify the behavior of McN-A-343 recently turned to the synthesis of truncated derivatives of the compound (44), with the aim of ascertaining whether distinct chemical moieties govern allosteric versus orthosteric actions of the compound. Figure 5⇓ represents minimal candidates for allosteric (DDBL-4) and orthosteric (TMA) pharmacophores; DDBL-4 has a substantial negative allosteric effect on the signaling efficacy of TMA, akin to the weak partial agonism displayed by the parent compound, McN-A-343, itself. In contrast to these actions at the M2 receptor, there is a minimal effect at the M1 muscarinic receptor where, presumably, the allosteric site is different between the two subtypes. Moreover, mutation of a key tyrosine residue to alanine (Y177A) in the second extracellular loop of the M2 muscarinic receptor, which is known to play a role in the potency of allosteric modulation, results in a significant blunting of the negative allosteric effect of DDBL-4 on TMA and a substantial increase in the apparent efficacy of McN- A-343 (Figure 5B⇓). In Figure 6⇓, a molecular model of the M2 muscarinic receptor, based on the crystal structure of the inactive β2 adrenergic receptor, suggests a possible bitopic binding mode for McN-A-343. The only substantial predicted interaction within the orthosteric pocket is that between the ammonium headgroup and a key aspartic acid (purple) in transmembrane domain 3, which is well-known to play a key role in the binding and activation mechanism of classic biogenic amines. Much of the molecule is predicted to sit high up in the receptor, towards the extracellular loop regions where Y177 (blue) resides, and may thereby engage the allosteric site.
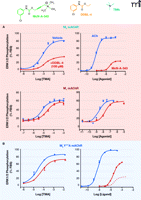
Functional selectivity as a consequence of a bitopic mechanism. Combination of the allosteric modulator DDBL-4 and orthosteric agonist TMA recapitulates the pharmacology of McN-A-343, a functionally selective muscarinic receptor agonist. (A) Effects of the indicated ligands on M2 or M1 muscarinic receptor–mediated ERK1/2 phosphorylation. DDBL-4 is a negative allosteric modulator of TMA efficacy at the M2 receptor but has a minimal effect at the M1 receptor, presumably due to differences between allosteric sites. Data for the M2 receptor are adapted from (44). (B) Mutation within the prototypical allosteric site of the M2 receptor reduces the potency of DDBL-4 as a negative efficacy modulator and increases the efficacy of McN-A-343; the dashed line indicates agonist behavior at the wild-type M2 receptor. Data are adapted from (44) (FBS, fetal bovine serum.)
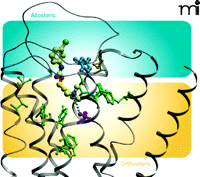
Molecular model of McN-A-343 docked in a bitopic mode to the M2muscarinic receptor. Residues in green indicate orthosteric site amino acids that do not affect the binding of McN-A-343. The purple residue is a key orthosteric aspartic acid (D103) in TM3 that acts as a counter ion for the ammonium headgroup. The blue tyrosine residue (Y177), in the allosteric pocket, affects efficacy of McN-A-343. See text for details.
The findings with McN-A-343 have a number of important implications. For instance, it is possible that recently identified, functionally selective muscarinic receptor agonists, such as AC-42, 77-LH-28-1, N-desmethylclozapine, and TBPB, may be bitopic ligands; interaction and/or mutagenesis studies have been presented to suggest that each of these molecules can bind allosterically (48, 51–56). Moreover, this situation may extend to other GPCRs; catechol, a key moiety within all catecholamines, can occupy the β2 adrenergic receptor concomitantly with the orthosteric inverse agonist ICI118551 (57), suggesting that a component of one “orthosteric” compound can act allosterically relative to a second orthosteric ligand. A second implication occurs with respect to GPCR ligand classification. Given that the identification of allosteric agonists is becoming more prevalent (29), it is important to ascertain whether such compounds are purely allosteric, or whether they also function directly at the orthosteric site. It is also possible that some bitopic ligands may not engage both orthosteric and allosteric sites concomitantly, as shown in Figure 3A⇑. Clearly, there are a number of possible variations of the bitopic theme, and future studies of the phenomenon will require a combination of equilibrium, kinetic, and mutagenesis experiments to validate particular mechanisms (41, 43, 44).
Conclusions
Despite being a relatively new concept, the conjugation of orthosteric ligand to allosteric modulator to yield a bitopic ligand is a logical extension of design approaches that have resulted in selective and/or potent GPCR-targeting ligands. The extent to which such compounds can be developed clinically remains to be determined, especially for large molecules for which “drugability” becomes an issue. At the very least, however, bitopic ligands clearly represent a new route towards engendering GPCR functional selectivity and, as tools, are likely to yield new insights into mechanisms of GPCR action.

Acknowledgments
Work in the authors’ laboratory is funded by National Health and Medical Research Council (NHMRC) of Australia Project Grant No. 400134 and Program Grant 519461, and Australian Research Council Discovery Grant No. DP0877497. AC is a Senior, and PMS a Principal, Research Fellow of the NHMRC.
- © American Society for Pharmacology and Experimental Theraputics 2009
References

Arthur Christopoulos, B.Pharm., PhD, is currently Professor of Pharmacology, a Senior Research Fellow of the National Health and Medical Research Council of Australia, and co-Director of the Drug Discovery Biology Laboratory in the Monash Institute of Pharmaceutical Sciences, Monash University, Melbourne, Australia. He is one of the leading international authorities on the study of small molecule GPCR allosteric modulators as a novel approach to GPCR drug discovery. His leadership responsibilities have included membership of seven international editorial boards, numerous consultancies and industry contracts, and he is currently a Corresponding Member of the NC-IUPHAR. Arthur’s other achievements include one international and five national science awards, over 90 invited international and national presentations, and over 100 publications. When not thinking allosterically, Arthur collects (and plays) guitars. Send correspondence to AC. E-mail arthur.christopoulos{at}med.monash.edu.au; fax +613 9905 5953.

Patrick M. Sexton, BSc (Hons) PhD, is currently Professor of Pharmacology, a Principal Research Fellow of the National Health and Medical Research Council of Australia, and co-Director of the Drug Discovery Biology Laboratory in the Monash Institute of Pharmaceutical Sciences, Monash University, Melbourne, Australia. Patrick is a leading international authority in the field of Family B GPCRs and on the interaction of GPCRs with accessory proteins. His leadership responsibilities have included membership of eight international editorial boards and an IUPHAR nomenclature subcommittee. He has been a chief investigator on over 50 research applications funded by both government and industry bodies. He has delivered over 100 invited international and national presentations and is an author of over 100 peer reviewed international journal articles.

Celine Valant, PhD, obtained her doctorate in Medicinal Chemistry from the University of Pharmacy in Strasbourg, France, under the supervision of Prof. Marcel Hibert. Her doctoral work focussed on the synthesis of receptor conformation state-specific allosteric inhibitors of the tachykinin NK2 receptor and fluorescently-labeled derivatives of muscarinic receptor modulators. She is currently a postdoctoral research fellow in the laboratory of Profs. Arthur Christopoulos and Patrick Sexton at the Monash Institute of Pharmaceutical Sciences, Monash University, Melbourne, Australia. Her current projects involve the investigation of structure-function and structure-activity relationships of various Family A GPCR modulators, including those of the adenosine A1 receptor and the M2 and M4 muscarinic receptors. During her time as a postdoc in Australia, her once outstanding golf handicap has deteriorated appreciably.

