Mechanisms of Disulfiram-induced Cocaine Abstinence: Antabuse and Cocaine Relapse
Abstract
Chemokines are chemoattractant proteins, secreted by a great many cell types, that function particularly to direct the migration of cells of the immune system. Chemokine receptors are G protein–coupled and have been implicated in a number of important disease states, including cancer, inflammatory disorders, and AIDS/HIV. Neuroinflammatory aspects of chronic pain are also chemokine-dependent, and given the many clinical difficulties that often arise in the treatment of chronic pain, novel therapeutics are urgently needed. Chemokine receptors would thus appear to be in ideal target for drug development, and indeed, animal models of neuropathic pain have allowed pharmacologists to identify a number of cell types and signaling pathways that are essential to the initiation and maintenance of chronic pain. In this regard, the chemokine receptor CCR2, activated by a chemokine known as monocyte chemoattractant protein 1, has been attributed to a number of specific regulatory and neural activation processes that contribute to chronic pain. But in order to target chemokine receptors such as CCR2 and thereby develop effective treatment, investigators must identify, from among many cell types and peripheral and central neural pathways, the relevant sites of CCR2 function.
Introduction
Disulfiram (Antabuse) first received pharmacological interest in the 1930s, after workers in a rubber factory, where the compound was used as an antioxidant, became ill. In particular, workers who consumed alcohol after having been exposed to disulfiram experienced flushing of the face, nausea, vertigo, headache, and hypotension. This series of unpleasant symptoms, now known as the “disulfiram-ethanol reaction” (1), eventually led, in 1951, to the approval of disulfiram, under the name Antabuse, by the Food and Drug Administration for the treatment of alcoholism. The drug’s efficacy relies on aversive conditioning; simply put, alcoholics who are prescribed Antabuse learn to avoid alcohol in order to avoid the negative consequences of the disulfiram-alcohol reaction. Today, there is a growing body of evidence to suggest that disulfiram also reduces cocaine use in dependent individuals, regardless of whether they abuse alcohol (2–4).
In this review, we discuss several pharmacological targets of disulfiram, including those involved in cocaine metabolism and catecholamine synthesis, with a focus on dopamine β-hydroxylase (DBH), which catalyzes the conversion of dopamine to norepinephrine and thereby controls the norepinephrine-to-dopamine ratio in central noradrenergic neurons (5). We also review the role of norepinephrine signaling in reward and drug-seeking behavior, and we propose an integrated mechanism to account for disulfiram-induced cocaine abstinence.
Disulfiram and Ethanol Metabolism
Ethanol is converted to acetaldehyde by the enzyme alcohol dehydrogenase, and acetaldehyde is further metabolized to acetate by aldehyde dehydrogenase (6). Disulfiram is an inhibitor of aldehyde dehydrogenase, which is directly relevant to its role in curbing alcohol consumption (Figure 1). The high levels of acetaldehyde that accumulate following alcohol ingestion in patients taking disulfiram cause the mild to moderate levels of facial flushing, weakness, throbbing headache, nausea, vomiting, sweating, vertigo, hypotension, and other unpleasant symptoms that typify the disulfiram-ethanol reaction (also known as the Antabuse reaction) (7, 8). The direct association of this aversive reaction with alcohol consumption establishes a psychological deterrent in alcoholic patients who abide by the dosing regimen. It is important to note here that, in addition to its effects on aldehyde dehydrogenase, disulfiram has many other targets. Upon absorption, disulfiram is immediately reduced to diethyldithiocarbamate (DDC) when it reacts with thiol groups (9). This metabolite of disulfiram is a potent copper chelator (10), and it can thereby affect the activity of copper-dependent enzymes such as monooxygenases, amine oxidase, cytochrome oxidase, microsomal carboxylesterase, and plasma cholinesterase.
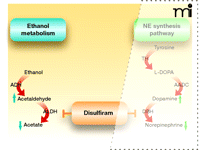
Disulfiram inhibition of ethanol metabolism. Ethanol is first converted into acetaldehyde by alcohol dehydrogenase (ADH). Acetaldehyde is then transformed into acetate by aldehyde dehydrogenase (ALDH). Disulfiram inhibits ALDH and thereby results in "the disulfiram-ethanol reaction" that promotes abstinence from alcohol. See text for details.
To date, there have been eight supervised clinical trials, ranging from 56 to 270 days in duration, that assess oral disulfiram for the treatment of alcoholics [see (11)]. The percentage of disulfiram-treated patients who completed these trials, ranging from eighteen to eighty-five percent, was higher for those populations in which drug administration was supervised by clinic staff or a family member. Only one study (comprising four disulfiram-treated patients and one placebo control) reported adverse side effects as a factor in patient drop-out (12). Four trials had a completely randomized design; three of these compared supervised versus unsupervised disulfiram administration (13–15). All groups reported better abstinence from drinking after supervised disulfiram administration compared with unsupervised administration or placebo. The studies of Robichaud et al. (16) and Sereny et al. (17), both using a within-subject design, found that disulfiram treatment led to alcohol abstinence in ninety-nine percent of patients during a 14-day treatment period, with fifty-eight percent of patients reaching “significant periods of sobriety.” In a six-month study (18), patterns of compliance within a supervised treatment program revealed that forty-five percent of subjects who took disulfiram regularly remained abstinent for the duration, whereas twenty-four percent of treated subjects relapsed. A recent investigation of 180 alcoholic patients randomly assigned to placebo or administration of the glutamatergic modulator acamprosate included a subset of subjects who volunteered to receive disulfiram (19). The simultaneous administration of acamprosate and disulfiram doubled the “mean cumulative abstinence” score in patients compared with the placebo-alone group.
Adherence to treatment must be recognized as a confounding factor in interpreting the effectiveness of disulfiram from clinical trial data. Problems with adherence pose the main clinical challenge in using disulfiram to treat alcoholism. Other limitations of its use are side effects (20–22) and hepatic toxicity in alcoholics with compromised liver function (10, 23), both of which are a result of disulfiram’s multiple enzymatic targets and lead to the underuse of the drug as a pharmacotherapy for alcoholism. Disulfiram can be an effective treatment for alcoholism, especially when patient compliance is supervised or supported within the context of stable social circles and community ties. It is against this backdrop that the idea of treating cocaine dependence with disulfiram emerged in the early 1990s.
Disulfiram and Cocaine Addiction
Cocaine and Alcohol Co-abuse
According to clinical and epidemiological studies, fifty to ninety percent of patients who abuse cocaine also abuse alcohol (24–27), and several factors could account for this comorbidity. Not only is there evidence that ethanol inhibits cocaine metabolism, but in addition, the simultaneous administration of the two drugs results in the formation of the active metabolite cocaethylene (CE) (28–32). CE manifests a pharmacological profile similar to that of cocaine in a number of respects: it has a high affinity for the dopamine transporter; it increases extracellular dopamine concentrations (33–35); it decreases the firing rate of dopamine neurons in the ventral tegmental area (VTA); and it potentiates ethanol-induced excitation of VTA neurons (36). In contrast to cocaine, however, CE is less potent in elevating heart rate and has a longer half-life (37). In the rat, CE is more reinforcing and less anxiogenic than cocaine (38), and CE overdose is more lethal than an overdose of either cocaine or ethanol alone (39). Taken together, these observations may explain the frequent anecdotal accounts from cocaine users who report that alcohol use prolongs their cocaine-induced euphoria (“high”) and reduces their paranoia during a cocaine binge. Similarly, alcohol reportedly helps “take the edge off” of cocaine-induced anxiety, hyperactivity, and insomnia. And finally, alcohol intoxication can impair judgment and inhibition, which may lead to further risky behavior, including illicit drug use.
Disulfiram Treatment for Dual Cocaine and Alcohol Dependence: Clinical Trials
On the hypothesis that withholding alcohol from cocaine-and-alcohol–dependent individuals might lead to a decrease in cocaine use, two research groups examined the use of disulfiram in this patient population in 1993. In the first study, although disulfiram treatment reduced both alcohol and cocaine use, the effect on cocaine use was attributed to a course of behavioral therapy that had been implemented in the patient population (40). The second study, a randomized twelve-week pilot trial, compared the effects of disulfiram administration to the effects of naltrexone, an opioid antagonist that may prevent drug craving; both treatments were accompanied by cognitive behavioral therapy. Disulfiram proved more effective than naltrexone at lowering the frequency of cocaine use (41), and a larger clinical trial ensued, in which disulfiram treatment improved abstinence from cocaine as compared to no treatment; disulfiram treatment in this larger trial appeared more effective for those outpatients who also received cognitive behavioral therapy (CBT) (42). The beneficial effect of disulfiram was still evident a year later in a follow-up study (43), and the efficacy of disulfiram therapy in diminishing cocaine use as observed earlier in comorbid addicts (41) was corroborated.
The notion that the mechanism of disulfiram-induced cocaine abstinence might not be related to the disulfiram-alcohol reaction emerged from the results of two studies published in 2000. In these trials, the effects of disulfiram on cocaine use were assessed in patients who were addicted to both cocaine and opiates, and who were maintained on methadone (3) or buprenorphine (2). In agreement with previous studies, the addicts treated with disulfiram were better able, relative to those addicts not receiving disulfiram, to reduce their intake of alcohol, cocaine, and opiates (3). In addition, disulfiram shortened the time necessary for patients to reach continuous cocaine, but not heroin, abstinence (2). Throughout these trials, alcohol consumption was minimal for all subjects, regardless of medication group, and baseline alcohol use did not predict responses to disulfiram. It was therefore something of a conceptual breakthrough when, in 2004, a randomized, placebo-controlled trial not only confirmed the effectiveness of disulfiram in treating cocaine dependence, but moreover revealed that the drug’s effectiveness in this regard could be differentiated from its role in curbing alcohol abuse (4). Specifically, the groundbreaking trial addressed cocaine use both with and without comorbidity for alcohol abuse, showing that the benefits of disulfiram therapy were most pronounced in patients who either were not alcohol dependent at baseline or who fully abstained from alcohol during treatment. These observations directly suggest that disulfiram undermines cocaine addiction in a manner independent of its action in inhibiting alcohol intake. Several other double-blind, randomized, placebo-controlled trials have confirmed the efficacy of disulfiram on cocaine intake (44, 45), with a potentially greater effect in males (46), but none have been designed to investigate the mechanisms of disulfiram-induced cocaine abstinence. Nevertheless, some clues can be gleaned from human laboratory and animal studies assessing the interactions between disulfiram and the physiological, behavioral, and subjective effects of cocaine.
Disulfiram and Cocaine: Human Laboratory and Animal Studies
To understand how disulfiram treatment affects an individual’s response to cocaine, and to shed light on the mechanism driving the drop in cocaine intake, a series of human laboratory studies looked at whether disulfiram influences the self-reported subjective effects of cocaine. The results have been conflicting. Two groups reported no difference in the subjective effects of cocaine, such as cravings or high, using a “Yes/No” scale (3) or a visual analog scale (47). Another group reported a modest, non-significant increase in “high” and “anxiety” (48), whereas others found increases in nervousness, paranoia (49, 50), or psychosis (51, 52). Two other studies report decreased “rush,” “high,” or “craving” (44, 53). Disulfiram is also reported to enhance some subjective effects of the psychostimulant dextroamphetamine, including “high,” anxiety, “bad drug effects,” “craving,” and “drug liking” (54). The modulation of subjective effects by disulfiram may in fact vary, increasing or decreasing the rewarding effects of cocaine; the most consistent finding is a worsening of the aversive effects of cocaine, such as anxiety and paranoia. Other side effects of disulfiram clinical trials on cocaine dependence are headaches, fatigue, and paranoia (55). Thus, there may be a “disulfiram-cocaine” reaction that is similar to but distinct from the “disulfiram-alcohol” reaction and that promotes cocaine abstinence.
There are a few published preclinical studies in rodents that address how disulfiram pretreatment affects behavioral responses to cocaine. Early studies showed that disulfiram pretreatment suppresses amphetamine-induced (56) and cocaine-induced (57) locomotor activity in mice and rats. More recent studies indicate that disulfiram has minimal effects on baseline activity levels, but repeated administration prior to cocaine facilitates the development of behavioral sensitization to cocaine in rats (58) and mice (our unpublished data). Disulfiram pretreatment also enhances cocaine-induced seizures in mice (59).
Molecular Mechanisms of Disulfiram Action
Dopamine Metabolism and Release
Because dopamine mediates many of the addictive properties of cocaine and other psychostimulants, the effect of disulfiram on dopamine neurotransmission is a logical place to look for clues to the drug’s clinical efficacy. Although clinical trials have shown that the disulfiram-ethanol reaction cannot account for effects on cocaine dependence, aldehyde dehydrogenase in fact plays two important roles in dopamine metabolism. Dopamine is metabolized by monoamine oxidase into the intermediate metabolite 3,4-dihydroxyphenylacetaldehyde (DOPAL), which is then converted by aldehyde dehydrogenase into 3,4-dihydroxypenylacetic acid (DOPAC) (60). DOPAL is a reactive electrophile and is toxic to dopaminergic neurons (61). In a separate metabolic pathway, dopamine is converted by catechol-O-methyl-transferase and monoamine oxidase to produce 3-methoxy-4-hydroxyphenylac-etaldehyde (MHPA), which is a substrate of aldehyde dehydrogenase. In this way, disulfiram might be expected to increase levels of both DOPAL and MHPA (Figure 2). Because the accumulation of DOPAL decreases dopamine uptake into synaptic vesicles and is toxic to dopamine neurons, disulfiram might be predicted to undermine dopamine transmission and dampen the euphoric and stimulant effects of cocaine. However, patients in some laboratory studies report just the opposite, namely, augmentation of the “high” elicited from cocaine (48) and dextroamphetamine (54); these results are inconsistent with the inhibition of aldehyde dehydrogenase as a basis for disulfiram-induced cocaine abstinence.
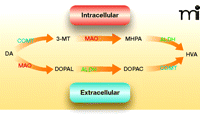
Dopamine metabolism. Dopamine (DA) is metabolized intracellularly and extracellularly by the same group of enzymes, but in different orders. Inside dopaminergic cells, monoamine oxidase (MAO) converts dopamine into 3,4-dihydrophenylacetaldeyde (DOPAL), a substrate of aldehyde dehydrogenase (ALDH). ALDH then converts DOPAL into 3,4-dihydroxyphenylacetic acid (DOPAC). After DOPAC diffuses out of the cell, catechol-O-methyltransferase (COMT) converts it into homovanillic acid (HVA). Extracellularly, dopamine metabolism begins by transformation into 3-methoxytyramine (3-MT) by COMT. 3-MT is then oxidized into 3-methoxy-4-hydroxyphenylacetaldehyde (MHPA) by MAO, and finally transformed into HVA by ALDH.
There has been one microdialysis investigation into the effects of disulfiram upon the storage and release of striatal dopamine and DOPAC in rats (62). In this study, disulfiram was reported to increase dopamine and DOPAC release in the striatum; however, the acute systemic dose (500 mg/kg) was notably higher than behaviorally relevant doses administered in previous studies (56–58, 63–66). The observed increase in dopamine release (145% above baseline level), moreover, peaked at forty minutes after treatment and returned to baseline after eighty minutes, whereas lower (behaviorally relevant) disulfiram dosing has been observed by ourselves (unpublished data) and others (65, 66) to elevate tissue dopamine concentrations for hours following administration. In vitro indications that disulfiram inhibits uptake and increases efflux of dopamine in bovine striatal synaptic vesicles (62) are similarly problematic, because the concentration of disulfiram used (1.7 mM) was well beyond physiologically relevant levels. Nevertheless, the data indicate that disulfiram can directly disrupt dopamine metabolism and homeostasis. There is not sufficient evidence, however, to support the conclusion that such disruption can account for the drug’s efficacy in promoting cocaine abstinence. It is, rather, an indirect effect upon dopamine release that likely accounts for the therapeutic effect of disulfiram (see below).
Glutamate Neurotransmission
Glutamate is the primary excitatory neurotransmitter in the mammalian central nervous system. Brain regions that mediate the addictive properties of cocaine, such as the mesolimbic dopamine system (VTA and nucleus accumbens), amygdala, and frontal cortex, all receive extensive glutamatergic projections [see (67) and (68)]. Given this anatomical correlation, as well as the functional correlation between dopamine and glutamate in learning and memory, much recent research has focused on dopamine-glutamate interactions in the modulation of psychostimulant-induced synaptic plasticity and addiction (69–71). Pharmacological manipulation of glutamate receptors can have profound effects on behavioral responses to psychostimulants in animal models of addiction. For example, ionotropic and metabotropic glutamate receptor antagonism impairs the acquisition and expression of cocaine-conditioned place preference (72, 73) and attenuates cue-induced cocaine seeking in rats (74–79).
The effects of disulfiram on glutamatergic neurotransmission are not well characterized. One study showed that S-methyl-N,N-diethylthiocarbamate sulfoxide (DETC-MeSO), a metabolite of disulfiram, blocked glutamate binding to receptors in mouse brains (80). The blockade was dose-and time-dependent partially (up to 50%) irreversible, and affected more than one receptor subtype. Another group reported that disulfiram, but not its primary metabolite DDC, attenuated synaptic glutamate uptake in rat brain and in cultured neurons, resulting in higher extracellular levels of glutamate (81). Still another study showed that both disulfiram and DDC increased extracellular glutamate levels in the rat striatum (82); here, DCC (290 mg/kg) elicited a brief two-fold increase in glutamate, whereas disulfiram (500 mg/kg) was less potent but longer-lasting. The interpretation of this microdialysis study is limited by the high doses of drug used. Furthermore, extracellular levels of glutamate during cocaine withdrawal differ among brain areas, and there are disulfiram interactions with cocaine in this regard. For example, during cocaine withdrawal, glutamate is decreased in the nucleus accumbens (83–85) but increased in the prefrontal cortex (86). Although normalizing glutamatergic transmission in the nucleus accumbens might therefore seem like a promising therapeutic strategy for treating psychostimulant addiction (87), there is currently insufficient evidence to support a critical role for altered glutamatergic transmission in the therapeutic efficacy of disulfiram.
Cocaine Metabolism
Disulfiram is an inhibitor of plasma cholinesterase and microsomal carboxylesterase activities (88), both of which are essential for cocaine metabolism (89, 90). This inhibitory activity likely explains the reported three-to sixfold increase in plasma cocaine levels following intranasal administration of disulfiram (47–49). In one study (48), serum cholinesterase activity was surprisingly not affected by disulfiram treatment; however, the authors acknowledged the limitations of the assay used and their sample preparation, so that cholinesterase inhibition may have been present but undetected. We observed no effect of disulfiram on peak serum cocaine levels in wild-type mice despite potent behavioral effects (59). In theory, increases in plasma cocaine levels in disulfiram-treated addicts could enhance both rewarding and aversive responses to cocaine. Nonetheless, addicts are quite adept at titrating their doses to maximize euphoric and minimize unpleasant effects and could thereby circumvent the abstinence-promoting effects of treatment.
Dopamine β-Hydroxylase Inhibition
Although any of the possible mechanisms described above could contribute to disulfiram-induced cocaine abstinence, none of them are especially compelling, and some are difficult to test. In contrast, data from animal and human laboratory studies strongly support an important role for DBH inhibition in the ability of disulfiram to reduce cocaine use. The following section will review and integrate the available data and suggest further testable hypotheses concerning DBH inhibition and the blockade of norepinephrine synthesis as primary mechanisms underlying disulfiram-induced cocaine abstinence.
Cocaine inhibits the reuptake of dopamine, serotonin, and norepinephrine by transporters at the plasma membrane of mono-aminergic neurons and thus elevates extracellular concentrations of these neurotransmitters (91–93). The psychostimulant effects of cocaine are mediated primarily by enhancement of dopaminergic transmission in the mesolimbic system in the brain, while serotonin and norepinephrine play a modulatory role. Based on how these neurotransmitters mediate drug responses and reward, most pharmacotherapeutic strategies to combat cocaine addiction have focused on the modulation of dopamine signaling.
DBH converts dopamine to norepinephrine, thereby playing a direct role in determining the ratio of dopamine to norepinephrine in noradrenergic neurons (64, 94). The enzyme is copper-dependent (95), and because the primary metabolite of disulfiram, DDC, is a copper chelator, DBH activity is inhibited by disulfiram (65, 96). Disulfiram administration decreases central conversion of dopamine into norepinephrine and significantly lowers the ratio of norepinephrine to dopamine in the brains of mice and rats (57, 64, 66, 97). (See Figure 3.)
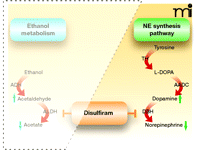
Disulfiram inhibition of the norepinephrine (NE) biosynthetic pathway. In the catecholamine synthesis pathway, tyrosine is converted into 3,4-dihydroxy-L-phenylalanine (L-DOPA) by tyrosine hydroxylase (TH), which is then transformed into dopamine by aromatic amino acid decarboxylase (AADC), whereupon dopamine b-hydroxylase (DBH) converts dopamine into norepinephrine. Disulfiram inhibits DBH, reducing the production of norepinephrine and increasing the pool of dopamine.
DBH Inhibition in Cocaine Reward, Aversion, and Relapse
We begin this section with three ways in which DBH inhibition has been hypothesized to decrease cocaine use: (1) by acting as a “dopamine replacement therapy;” (2) by decreasing the rewarding effects of cocaine; and (3) by increasing the aversive effects of cocaine. Ultimately, however, we will argue that relapse prevention mediated by decreased levels of norepinephrine is likely the most important mechanism underlying disulfiram-induced cocaine abstinence.
The “Dopamine Agonist” Hypothesis of DBH Inhibition
Chronic cocaine users are hypothesized to be hypodopaminergic, and a popular treatment strategy has been to “normalize” dopaminergic tone [e.g., dopamine agonist therapy; reviewed by Grabowski et al. (98)]. Indeed, disulfiram has sometimes been classified as a “dopamine agonist,” as its beneficial effect on cocaine intake has been attributed to increased dopamine concentration following DBH inhibition (99). However, we will argue that, because of the functional interaction between the noradrenergic and dopaminergic systems, this hypothesis is unlikely to be correct.
Anatomical and functional connections between the noradrenergic and dopaminergic systems affect the responsiveness of the mesolimbic reward system to psychostimulants. The locus coeruleus, the primary noradrenergic nucleus in the brain, directly innervates the VTA and the prefrontal cortex. The A1 and A2 noradrenergic cell groups in the brain stem also project to the VTA and the nucleus accumbens. In general, norepinephrine (i.e., the product of DBH activity) provides excitatory drive onto midbrain dopamine neurons, both directly (through the VTA) and indirectly (via glutamatergic projections from the prefrontal cortex to the VTA) [reviewed by Weinshenker and Schroeder (100) and Mejías-Aponte et al. (101)]. In this way, DBH inhibition would decrease excitatory drive onto midbrain dopamine neurons, and thus, despite increased tissue levels of dopamine, the firing of dopaminergic neurons (particularly burst firing) would be attenuated, and extracellular DA levels would actually fall. For example, pharmacological blockade of norepinephrine signaling inhibits basal and psychostimulant-induced burst firing in midbrain dopaminergic neurons (102, 103), and lesions of the locus coeruleus, along with norepinephrine depletion, reduce dopamine release in terminal regions (104). Also, norepinephrine depletion in the prefrontal cortex attenuates psychostimulant-induced dopamine release in the nucleus accumbens (105, 106). Finally, although DBH knockout (Dbh−/−) mice completely lack norepinephrine and have high tissue dopamine levels, the animals manifest low basal and psychostimulant-evoked extracellular dopamine levels in the dorsal and ventral striatum (107). The specific effects of disulfiram on extracellular catecholamines have yet to be tested, but treatment of mice with another copper chelator (fusaric acid) also acts to inhibit DBH and diminishes basal and methamphetamine-provoked dopamine release in the striatum (108). These results suggest that extracellular dopamine levels are likely reduced, not raised, by disulfiram, effectively refuting the idea that disulfiram treatment promotes cocaine abstinence by “normalizing” dopamine levels in addicts (see Figure 4).
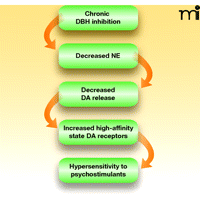
Effect of chronic DBH inhibition on dopamine transmission. Genetic DBH inhibition, and presumably pharmacological DBH inhibition by disulfiram, leads to decreased norepinephrine synthesis in the locus coeruleus and brainstem and norepinephrine release in the midbrain. Because midbrain dopaminergic neurons require noradrenergic drive for normal burst firing and neurotransmitter release, dopamine release is decreased and a compensatory upregulation of high-affinity state dopamine receptors ensues, resulting in behavioral hypersensitivity to psychostimulants.
Reduction in Cocaine-Induced Reward Effects by DBH Inhibition
Because noradrenergic facilitation of dopamine transmission is crucial for the rewarding effects of psychostimulants, disulfiram might be reasoned to reduce cocaine use by inhibiting the noradrenergic-mediated high. Indeed, norepinephrine depletion or adrenergic receptor blockade attenuates the stimulant and rewarding effects of amphetamine and cocaine in rodents [reviewed in (100)]. For example, norepinephrine promotes the locomotor-activating effects of psychostimulants and also facilitates dopamine release in the nucleus accumbens and conditioned place preference (105, 106, 109–112). Consistent with this hypothesis of DBH inhibition, one human laboratory study found that disulfiram decreased cocaine “high” and “rush” (53). On the other hand, several studies show no effect or even higher levels of psycho-stimulant reward following disulfiram administration (3, 47–49, 54). In any event, therapeutic strategies to blunt the rewarding effects of psychostimulants are unlikely to be successful, because drug addicts can easily defeat such measures by adjusting their drug intake. For instance, dopamine receptor antagonists actually tend to increase rather than decrease cocaine self-administration in rats (113–116). It is thus not reasonable to expect partial reduction of cocaine reward to significantly modify the behavior of cocaine-dependent individuals.
Cocaine Aversion via DBH Inhibition
The mesolimbic system and dopamine release are important not only for reward, but also for responses to aversive stimuli (117–119). Because norepinephrine depletion attenuates drug-induced dopamine release, as described above, DBH inhibition might also be expected to blunt the aversive effects of cocaine. Although this may be true for acute DBH inhibition, chronic DBH inhibition (as might be observed following days or weeks of disulfiram treatment) leads to compensatory changes in the dopaminergic system that result, paradoxically perhaps, in a hypersensitivity to the aversive effects of cocaine. A striking example of this phenomenon occurs in Dbh−/− mice, which essentially have total and lifelong DBH inhibition (120, 121). Given their low basal levels of dopamine release, which remains low in spite of stimulant administration, these mice manifest a compensatory increase in striatal high-affinity state dopamine receptors (107). Consequently, Dbh−/− mice have a paradoxical hypersensitivity to psychostimulants, manifested in heightened locomotor activity and related stereotypical behavior (107, 122). Even more relevant to this discussion is the observation that the same cocaine doses that evoke conditioned place preference in wild-type mice result in conditioned place aversion in Dbh−/− mice. The Dbh−/− phenotype thus suggests that chronic norepinephrine deficiency augments the aversive effects of cocaine.
The augmentation of aversive behavior in Dbh−/− mice may offer an explanation for similar behavior seen in people. Plasma DBH activity is highly variable in humans, and a significant proportion of this variability is genetic (123). A common polymorphism, a C-to-T change at nucleotide position -1021 of the DBH gene (allele frequency ~0.2) accounts for most of the genetic variance in DBH activity; individuals heterozygous for the low-activity T allele (i.e., individuals who are “CT”) have approximately 50% lower DBH activity than CC homozygotes; DBH activity in TT homozygotes is typically reduced by greater than 90% relative to that of CC individuals (124). Strikingly, cocaine addicts with genetically low DBH activity report increased cocaine-induced paranoia (125, 126), an effect that is phenocopied by disulfiram treatment prior to cocaine administration [R. Malison, personal communication; see also (49, 50, 127)].
Could an increase in the aversive effects of cocaine, such as paranoia, underlie disulfiram-induced cocaine abstinence? The success of disulfiram in the treatment of alcoholism depends on just such a negative response to the addictive drug. In the case of ethanol, inhibition of aldehyde dehydrogenase produces an aversive reaction mediated by acetaldehyde. Perhaps in the case of cocaine, chronic inhibition of DBH produces an aversive reaction mediated by the induced expression of high-affinity dopamine receptors and excessive dopamine signaling. Appealing as this hypothesis may be, however, increased cocaine aversion is probably not a major contributor to the clinical efficacy of disulfiram. As discussed above, cocaine addicts habitually titrate their cocaine intake to achieve the desired subjective effects. If high doses of cocaine were producing a negative experience, they would probably lower their intake, not abstain. In fact, at low doses of cocaine, Dbh−/− mice actually seem to experience increased reward instead of aversion (107). Furthermore, neither adrenergic receptor antagonists nor disulfiram affect operant cocaine self-administration in rats (128–130). Finally, if cocaine aversion induced by chronic DBH inhibition discouraged cocaine use, one would predict that low-activity DBH alleles would be underrepresented in addicts, a prediction that is refuted by current data (131).
In summary, DBH inhibition is by far the most likely mechanism underlying disulfiram-induced cocaine abstinence for several reasons: i) the inhibition of DBH alters neurotransmitter levels known to be critical for the rewarding, aversive, and addictive properties of cocaine; ii) both mice and humans with low DBH activity have altered responses to cocaine; and iii) the altered responses to cocaine in DBH-deficient animals can be mimicked by disulfiram treatment. Interestingly, a pharmacogenetic interaction between disulfiram and DBH may exist; disulfiram appears to lower cocaine use primarily in individuals carrying at least one low-activity DBH allele (132, 133). This intriguing finding reinforces the idea that DBH inhibition contributes to the therapeutic effects of disulfiram in the treatment of cocaine dependence. However, all the potential hypotheses to account for why DBH inhibition is effective (e.g., dopamine receptor agonism, decreased cocaine reward, and increased cocaine aversion) fail upon a reasoned consideration of current data. In the final section, we will argue that relapse prevention, mediated by DBH inhibition and a decrease in norepinephrine, provides the most likely mechanistic explanation.
Norepinephrine and Reinstatement
Drug addiction is a chronic relapsing disorder (134, 135), as patients in treatment often slip back into drug-taking behaviors after periods of sobriety. Several types of stimuli can trigger drug craving and lead to relapse. Environmental and sensory cues associated with past drug use can prompt strong urges to renew drug use, an occurrence known as cue-induced relapse (136–138). Re-exposure to illicit drugs can also lead to reinstatement of drug abuse, which is called drug-primed relapse. Stress is the classic trigger for drug use, in the initiation of drug use, and the maintenance of addictive behavior (137, 139). Anecdotally, stress is cited as the most common cause of relapse. The organism’s natural reward system is neurologically usurped in addiction (140, 141), and so it is crucial that pharmacotherapies for cocaine addiction be selective in repressing the drive to seek drugs without altering the healthy impulse to seek out natural rewards. Prevention of relapse is thus considered one of the most promising strategies for current pharmacotherapies targeted at cocaine dependence; such strategies are based on efforts to dissociate drug-related cues and the effects of stress from drug use.
Although pharmacological manipulation of the noradrenergic system does not affect cocaine self-administration in rats or non-human primates, it profoundly affects the reinstatement of cocaine seeking after a period of abstinence or extinction of the behavior, and the effect on reinstatement in experimental mammals has become a commonly used model of human relapse. In such experiments, animals are first conditioned to self-administer a drug by pressing a lever that results in drug infusion, and then they are exposed to an “extinction” phase, during which responses on the active lever result only in saline infusions. During extinction, cues previously associated with drug delivery (i.e., tone and illumination of cue light) are absent. Following the extinction phase, “reinstatement” tests begin, during which rats are exposed to a relapse trigger, such as an injection of drug, acute stress (e.g., foot shock), or environmental cues associated with drug delivery (i.e., tone and light). These tests are referred to as drug-primed, stress-induced, and cue-induced reinstatement, respectively. Reinstatement of drug seeking is operationally defined as the number of responses on the active lever that previously resulted in cocaine infusion, even though the animals are under extinction conditions (i.e., still receiving saline infusions). The validity of reinstatement tests to model human addiction is high, as determined in studies of both humans and laboratory animals (142).
Studies have shown that reduction of norepinephrine signaling attenuates reinstatement of amphetamine- (143) and cocaine-seeking, although it appears that this attenuation can be mediated by different receptors; blockade of β-adrenergic receptors prevents stress-induced reinstatement, whereas blockade of α1-adrenergic receptors abrogates drug-primed reinstatement (144, 145). Preliminary results indicate that the blockade of cocaine-primed reinstatement by α1-adrenergic receptor antagonists can be mimicked by disulfiram (our unpublished data). Conversely, the facilitation of norepinephrine transmission (e.g., induces reinstatement in rats and non-human primates (146–148).
The contribution of norepinephrine to stress-induced reinstatement is particularly intriguing: emotional stress has been associated with increases in drug craving (149, 150); high stress is a good predictor of continued drug use in addicts (149); and individuals exposed to stress are more likely to relapse into drug use (150, 152). There is an overlap among the neurocircuits implicated in drug abuse and stress [see (153–155)]. The extended amygdala [i.e., bed nucleus of the stria terminalis, central amygdala, and the shell of the nucleus accumbens (156)], receives corticotropin-releasing factor and noradrenergic innervation, and this concerted innervation underlies the role of the extended amygdala in sensory information integration, emotion, and appetitive learning. Thus, it functions as a mediator of both stress and drug responses and integrates the function of norepinephrine in influencing stress-induced reinstatement [see (157)]. For example, local infusions of β-adrenergic receptor antagonists within the extended amygdala block stress-induced reinstatement of cocaine self-administration in rats (144). Finally, we and others have shown that β-adrenergic receptor antagonists attenuate cocaine-induced and cocaine withdrawal-induced anxiety (158, 159). Together, these results indicate that norepinephrine mediates stress responses that contribute to relapse. We propose that by decreasing noradrenergic transmission, disulfiram can dampen the stress response and lower the likelihood of relapse in the face of environmental influences and stressors.
Currently, there is little direct evidence supporting an effect of disulfiram on relapse prevention, as most clinical trials have focused on its effect on current cocaine use. To date, there has been only one study that measured outcomes in a group of patients for one year following monitored disulfiram treatment (43). Recorded outcomes in this follow-up study were complete abstinence and number of days of cocaine use (during the previous four weeks) self-reported at one, three, six, and twelve months after treatment completion. Frequency of drug use is a point-prevalence variable that gives a snapshot of a disease at a specific point in time. This measure is touted as a good reflection of the nature of drug use because it takes into account the fact that patients often relapse briefly into drug use and also allows for validation of self-reports with biochemical measures. Unfortunately, when this measure is not accompanied by thorough interviewing of the study participants, it fails to provide a broad view of the effect of a treatment. We speculate that if disulfiram primarily induces an increase in the aversive effects of cocaine, measures of frequency and amount of cocaine used would gradually decrease, as drug addicts would titrate drug intake to achieve the desired subjective effects. Conversely, if disulfiram primarily prevents relapse by blunting the drive to take drug following environmental stressors or cues, one might expect to observe a decrease in latency to abstinence and/or extended periods of abstinence. Carroll and colleagues reported that more study participants assigned to the disulfiram/CBT treatment group achieved complete abstinence from cocaine during the following year than those assigned to the placebo/CBT group. We urge that future clinical studies include interviews with participants and measures that can distinguish between abstinence due to altered subjective drug effects vs healthier responses to environmental triggers.
Conclusion
Disulfiram has been used as an alcohol deterrent for decades, and recent studies indicate that it is also an effective pharmacotherapy for the treatment of cocaine dependence; however, the mechanisms behind its efficacy for alcohol and for cocaine addiction are distinct. Whereas aldehyde dehydrogenase is the primary target in treating alcoholism, human laboratory, genetic, and preclinical animal studies indicate that its beneficial effects on cocaine use result from the inhibition of DBH. Despite the potential of DBH and its inhibition to modulate cocaine reward and aversion, we argue that the most important clinical effect of disulfiram-mediated DBH inhibition arises from the drug’s ability to reduce relapse, particularly, relapse precipitated by stress.
This hypothesis will require further studies in preclinical models of addiction. First, we must explore the effect of acute and chronic disulfiram treatment on several aspects of dopamine transmission, such as neurochemical and behavioral responses to psychostimulants. Second, we need to know the effects of disulfiram on stress-induced, cue-induced, and drug-primed reinstatement of cocaine seeking, as well as the brain region(s) critical for these effects. Third, disulfiram as a treatment for dependence on psychostimulants other than cocaine should also be investigated, as has been suggested by human laboratory studies of amphetamine-like drugs. Pharmacogenetic interactions between disulfiram and DBH genotype may also yield valuable information. Finally, the knowledge acquired by studying disulfiram could be translated into safer and more effective pharmacotherapies for the treatment of cocaine dependence. Because disulfiram use is limited by its non-specificity, side effects, and toxicity, the development and testing of selective DBH inhibitors will be essential to mechanistic studies as well as to improved therapeutics.

Acknowledgments
We thank Cheryl Strauss for assistance with text editing, Robert Malison for sharing unpublished results, and the National Institutes of Health and the National Institute of Drug Abuse for financial support. This work was supported by the National Institutes of Health National Institute of Drug Abuse [Grants DA017963, DA25040].
- Copyright © 2009
References

Meriem Gaval-Cruz, BS, is a graduate student in the Division of Biological and Biomedical Sciences at Emory University. Her research interests focus on the neurobiology of drug addiction, with an emphasis on cocaine pharmacotherapies. Her activities include neuroscience outreach to K-12 students and the laboratory mentoring of undergraduates.

David Weinshenker, PhD, is Associate Professor in the Department of Human Genetics at Emory University. He has pursued model systems to better understand genes involved in human disease. He applies genetic models combined with pharmacological tools to investigate various aspects of neurobiology, with a particular focus on the role of norepinephrine in brain function and disease. Send correspondence to DW. E-mail dweinshenker{at}genetics.emory.edu; fax 404-727-3949.

