|
 
 |
|
REVIEW ARTICLE |
|
|
|
| Year : 2011 | Volume
: 17
| Issue : 2 | Page : 48-53 |
| |
Craniosynostosis genetics: The mystery unfolds
Inusha Panigrahi
Department of Pediatrics, Genetic and Metabolic Unit, Advanced Pediatric Center, PGIMER, Chandigarh, India
| Date of Web Publication | 17-Oct-2011 |
Correspondence Address:
Inusha Panigrahi
Genetic and Metabolic Unit, Advanced Pediatric Center, PGIMER, Sec-12, Chandigarh
India
 Source of Support: None, Conflict of Interest: None  | 5 |
DOI: 10.4103/0971-6866.86171

 Abstract Abstract | | |
Craniosynsostosis syndromes exhibit considerable phenotypic and genetic heterogeneity. Sagittal synostosis is common form of isolated craniosynostosis. The sutures involved, the shape of the skull and associated malformations give a clue to the specific diagnosis. Crouzon syndrome is one of the most common of the craniosynostosis syndromes. Apert syndrome accounts for 4.5% of all craniosynostoses and is one of the most serious of these syndromes. Most syndromic craniosynostosis require multidisciplinary management. The following review provides a brief appraisal of the various genes involved in craniosynostosis syndromes, and an approach to diagnosis and genetic counseling.
Keywords: Apert syndrome, FGFR2 mutations, hydrocephalus, plagiocephaly, sutural synostosis, syndromes
How to cite this article:
Panigrahi I. Craniosynostosis genetics: The mystery unfolds. Indian J Hum Genet 2011;17:48-53 |
| Introduction | |  |
Craniosynostosis, premature suture fusion, is one of the most common craniofacial anomalies with incidence of 1 in 2,500 live births. Craniosynostosis can be isolated nonsyndromic or it may be part of a larger syndrome with digital malformations, skeletal defects, cardiac defect, or other organ anomalies. [1],[2] The sagittal suture is affected in 40-60% of cases, the coronal suture in 20-30% of cases, and the metopic suture in less than 10% of cases. More than 180 syndromes exist that contain craniosynostosis. The syndromes associated with craniosynostosis include Apert syndrome, Crouzon syndrome, Greig cephalopolysyndactyly, and Saethre-Chotzen syndrome. It is genetically heterogeneous disorder with mutation identified in several genes, predominantly the fibroblast growth factor receptor genes. [3],[4] Saethre-Chotzen syndrome and craniosynostosis (Boston-type) arise from mutations in the Twist and muscle segment homeobox 2 (MSX2) transcription factors, respectively. Rates of neuropsychological deficits range from 35 to 50% in school-aged children with isolated single suture craniosynostosis. [5] Secondary effects of craniosynostosis may include vision problems and increased intracranial pressure, among others. Patients with TWIST gene mutations may have more ophthalmic abnormalities, including more strabismus, ptosis, and amblyopia. [6] The following discussion gives a comprehensive review of different disorders presenting with craniosynostosis, their diagnosis, and genetic counseling.
| Clinical presentation of craniosynostosis syndromes | | |
- Isolated craniosynostosis: In this group, there is premature fusion of one or more of the sutures of the skull. This can be in the form of coronal synostosis. There are no associated skeletal defects, digital, or other anomalies.
- Apert syndrome: In classical Apert syndrome, there is brachycephaly, flat nasal bridge [Figure 1], and syndactyly of the fingers of hands called mitten hands, and the toes are also similarly affected. However, in nonclassical cases there can be variable syndactyly. [2],[4]
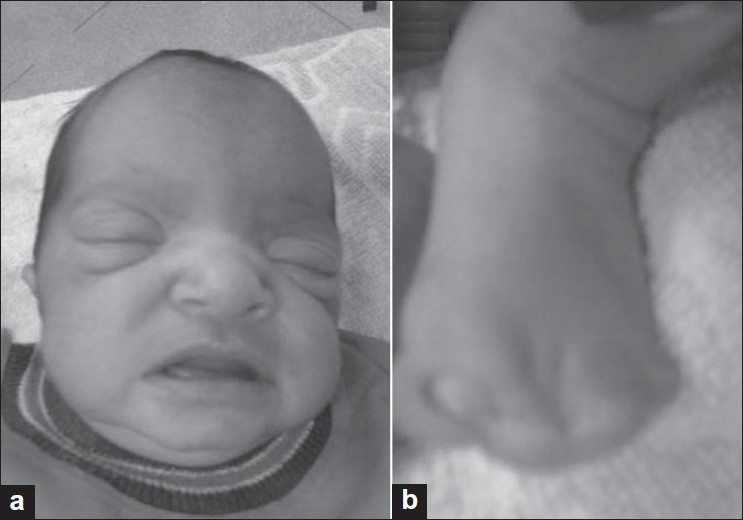 | Figure 1: Photograph of the face (a) in a case of Apert syndrome showing prominent forehead, hypertelorism, proptosis, low set ears, and open mouth. The child also had mitten hands. The feet with extensive syndactyly are shown in (b)
Click here to view |
- Crouzon syndrome: These patients usually have long face with proptosis, maxillary hypolasia, and a prominent jaw (mandibular prognathism). There is conductive hearing loss. It is associated with increased paternal age effect. The synostosis can involve the coronal, sagittal, and lambdoid sutures. [1],[2],[3] A variant of Crouzon syndrome is Crouzon syndrome with acanthosis nigricans (CAN) which is genetically different and there is progression of acanthosis with increasing age. The typical CAN patients present with craniosynostosis with crouzonoid faces, acanthosis nigricans with wide and atypical distribution, melanocytic nevi, choanal atresia or stenosis, hydrocephalus, Chiari malformation More Detailss, and oral abnormalities. [7],[8]
- Pfeiffer syndrome: The patients have hypertelorism, maxillary hypoplasia, mandibular prognathism, and turribrachycephaly. [1],[9] There is partial syndactyly of fingers and toes. There can be choanal atresia or stenosis or radiohumeral synostosis at elbows. Three types have been described: type 1-milder form; type 2-with cloverleaf skull and elbow ankylosis; and type 3-severe craniosynostosis, without cloverleaf skull, with early death.
- Cutis gyrata syndrome of Beare and Stevenson: Patients with this syndrome have midfacial hypoplasia, hypertelorism, and proptosis. The characteristic features are cutis gyrata involving the back of scalp, lumbosacral region, along with furrowed palms and soles; and acanthosis nigricans. [2],[10] There can be cloverleaf skull, hydrocephalus, developmental delay, and bifid scrotum. This is also associated with increased paternal age.
- Saethre-Chotzen syndrome: These patients have short stature, brachycephaly, acrocephaly, plagiocephaly, facial asymmetry, hypertelorism, beaked nose, deafness, and cardiac defect. [1],[4] There can be ptosis, buphthalmos, brachydactyly, syndactyly, clinodactyly, and radioulnar synostosis. Some patients have mild to moderate mental retardation [Figure 2].
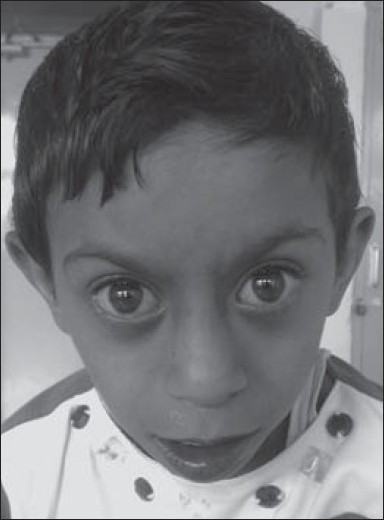 | Figure 2: Case clinically diagnosed as Saethre-Chotzen syndrome. She had short stature, mental retardation, triangular faces, prominent eyes, low set ears, short neck, brachydactyly, and cyanotic heart disease
Click here to view |
- Carpenter syndrome: There is brachycephaly with synostosis of coronal, lambdoid, and sagittal sutures; midface hypoplasia; low set ears; high arched palate; coxa valga; genu valgum; and polydactyly/syndactyly/clinodactyly/camptodactyly. [2],[11] Other features include conductive/sensorineural hearing loss, optic atrophy, cardiac defect (ASD/VSD/PS/TOF/PDA), and renal defects such as hydronephrosis and hydroureter.
- Muenke syndrome: The affected patients have macrocephaly, brachycephaly, plagiocephaly, with midfacial hypoplasia, coronal craniosynostosis, developmental delay, and sensorineural hearing loss. [12],[13] The acral anomalies include brachydactyly, clinodactyly, broad thimble like middle phalanges, broad toes, capitate-hamate fusions, and calcaneocuboidal fusions. Females are more severely affected than males. A majority of the patients (95%) show a mild-to-moderate, low frequency sensorineural hearing loss.
- Shprintzen-Goldberg craniosynostosis syndrome: The patients have dolichocephaly, prominent forehead, down slanting palpebral fissures, midfacial hypoplasia, hypertelorism, low set ears, and micrognathia. [14] There can be associated joint laxity or contractures, pectus excavatum or carinatum, scoliosis, camptodactyly, arachnodactyly, talipes equinovarus, umbilical, and inguinal hernias. The cardiac anomalies seen in this syndrome include aortic root dilatation and mitral valve prolapse.
- Baller-Gerold syndrome More Details: There is short stature, mental retardation, turribrachycephaly, hypertelorism, prominent nasal bridge, micrognathia, and low set ears. [1,2] The synostosis involves coronal, metopic, and lambdoid sutures. There can be associated cardiac defects, vertebral, and renal anomalies.
- Jackson-Weiss syndrome: These patients have craniosynostosis with midfacial hypoplasia, broad halluces with medial deviation, and cutaneous syndactyly of second and third toes. [1],[15]
- Craniosynostosis mental retardation syndrome of Lin and Gettig: The patients have fusion of sagittal, metopic, or lambdoid sutures leading to dolichocephaly, trigonocephaly, or turricephaly. There is mental retardation, midfacial hypoplasia, small nose, hypertelorism, ptosis, and blepharophimosis. Other abnormalities described are umbilical hernia, cryptorchidism, hydronephrosis, cardiac defects, and intestinal malrotation. [2]
- Hunter-McAlpine Craniosynostosis syndrome: There is short stature, mental retardation, facial dysmorphism with almond-shaped palpebral fissures and downturned corners of mouth, and subtle skeletal anomalies. [2]
- Antley-Bixler syndrome More Details (ABS): ABS is a multiple skeletal malformation syndrome with craniosynostosis, radiohumeral synostosis, femoral bowing, choanal atresia or stenosis, joint contractures, urogenital abnormalities, and, often, early death. [1],[16]
- POR (Cytochrome P450 Oxidoreductase) deficiency with Antley-Bixler phenotype: POR deficiency is a newly identified condition of congenital adrenal hyperplasia with or without skeletal anomalies. [16],[17] The presence of craniosynostosis, choanal atresia or stenosis, bowed femora, and radioulnar synostosis is suggestive of ABS phenotype.
- Opitz trigonocephaly syndrome: Partial or complete obliteration of the metopic suture is characteristic of this syndrome. The forehead is narrow and pointed, often associated with biparietal widening and triangular shape of the skull. There is facial dysmorphism with upslanting of the palpebral fissures, small nose with broad root, abnormally modeled ears, and short neck with loose skin. There may be polysyndactyly, omphalocele, or cardiac defect. [2]
- Craniofrontonasal dysplasia: Craniofrontonasal dysplasia is a rare, familial X-linked disorder with coronal synostosis (brachycephaly or plagiocephaly), hypertelorbitism (frequently asymmetric), and some extracranial anomalies such as scoliosis, congenital diaphragmatic hernia, and broad halluces. [18]
- Other craniosynostosis syndromes: Around 200 syndromes have been associated with craniosynostosis. [2],[19] Gomez-Lopez-Hernandez syndrome or cerebello-trigeminal-dermal dysplasia is a rare syndrome comprising short stature, cerebellar abnormalities, parieto-occipital alopecia, trigeminal nerve anesthesia, intellectual impairment, craniosynostosis, and craniofacial anomalies. Craniosynostosis-Boston type was described by Warman et al., in 1993. The characteristic features include forehead retrusion, frontal bossing, turribrachycephaly, and cloverleaf skull, with no hand/foot abnormalities. [4] Most affected individuals were myopic or hyperopic. Intelligence was normal. Other features described include seizures, short first metatarsal, and triphalangeal thumb. Craniosynostosis-Adelaide and Philadelphia types have also been described. Individuals with Adelaide-type craniosynostosis have various degrees of craniosynostosis leading to facial asymmetry, a broad forehead, brachyturricephaly, and facial abnormalities such as hypertelorism, maxillary hypoplasia, mandibular prognathism, a low anterior hairline, and hearing loss. Craniometaphyseal dysplasia manifests with macrocephaly, proptosis, hypocalcemia, hyperparathyroidism, wide metaphyses, and sensorineural deafness. Other conditions with craniosynostosis include Di-George syndrome, Smith-Magenis syndrome, triploid-diploid mosaicism, submicroscopic deletions of 11q25 and 9q22.3, camptomelic dysplasia, and Jansen metaphyseal dysplasia.
| Inheritance of craniosynostosis syndromes | | |
Many of the major syndromes have autosomal dominant inheritance or are sporadic. These include Crouzon syndrome, Apert syndrome, Pfeiffer syndrome, Saethre-Chotzen syndrome, and Jackson-Weiss syndrome. [1],[2],[3] The autosomal recessive disorders include Baller-Gerold syndrome, POR deficiency with ABS phenotype, Opitz C syndrome, and craniosynostosis-mental retardation syndrome of Lin and Gettig. Autosomal dominant forms of craniosynostosis-type Hoffmann and Mcgillivray have also been described.
| Genetic basis of craniosynostosis syndromes | | |
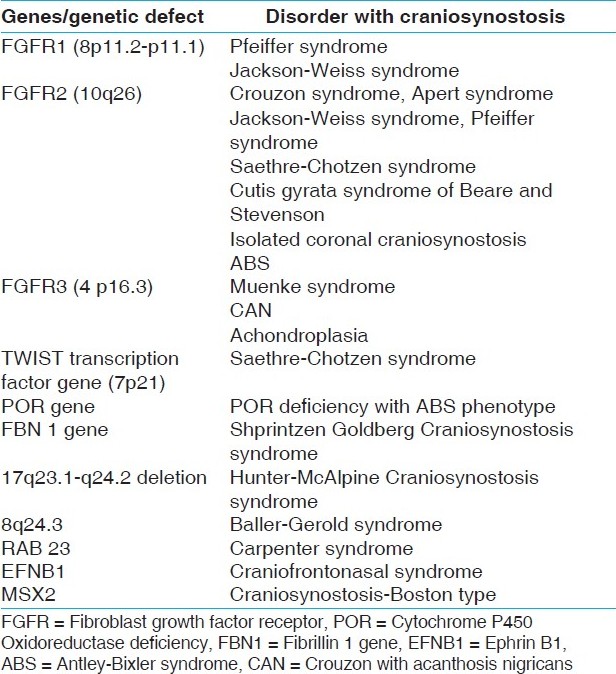
Recent advances in molecular genetics have led to a better understanding of the role of specific genes implicated in different craniosynostosis syndromes. [20],[21],[22],[23],[24] Most of these disorders or syndromes have mutations in the fibroblast growth factor receptor genes. These are FGFR 1, FGFR 2, and FGFR3. Mutations in the FGFR2, FGFR3, TWIST1, and EFNB1 genes have been shown to account for around 25% of craniosynostosis. Other genes implicated are the POR gene and FBN1 (Fibrillin 1) gene. Chromosomal alterations are important causative mechanisms of the syndromic forms of craniosynostosis accounting for at least 10% of the cases. [25],[26],[27] [Table 1] lists the different conditions along with underlying genetic defect. In craniosynostosis-type Mcgillivray, mutation in the TK1 portion of FGFR2 was found.
L1 cell adhesion molecule (L1CAM) gene plays a major role in the development of the white matter and its mutation in humans (callosal agenesis, retardation, adducted thumbs, spasticity, and hydrocephalus syndrome). A defect in interaction of FGFR with L1CAM may be the cause of the brain malformations and mental retardation in children with craniosynostosis. [28] Nonsyndromic craniosynostosis is a clinically and genetically heterogeneous condition that has the characteristics of a multifactorial trait.
| Genetic counseling | | |
The most common craniosynostosis syndromes being autosomal dominant in inheritance, the risk of recurrence is 50% for each pregnancy if there is one affected child or one parent affected. For autosomal recessive craniosynostosis syndromes such as Baller-Gerold syndrome, the recurrence risk is 25% in sibling. If only child is affected and there are no clinical features in parents or other family members, then the disease may be sporadic with negligible risk of recurrence.
| Management | | |
The pediatricians can identify the causes of the majority of abnormal head shapes by combining their understanding of normal calvarial growth with a careful physical examination [Figure 3]. The treatment of craniosynostosis syndromes-medical and surgical-is multidisciplinary and involves neurosurgeons, plastic surgeons, ENT specialist, pediatrician, and clinical genetics specialist. [29],[30] Craniofacial morphometry may help in planning surgery. [31] The high definition of three-dimensional computed tomography and magnetic imaging resonance allows precise surgery planning of reconstruction and management of associated malformations. [32] Midfacial surgery is performed to reduce the exophthalmos and the midfacial hypoplasia. An early surgery and team effort is necessary to optimize the long-term outcomes.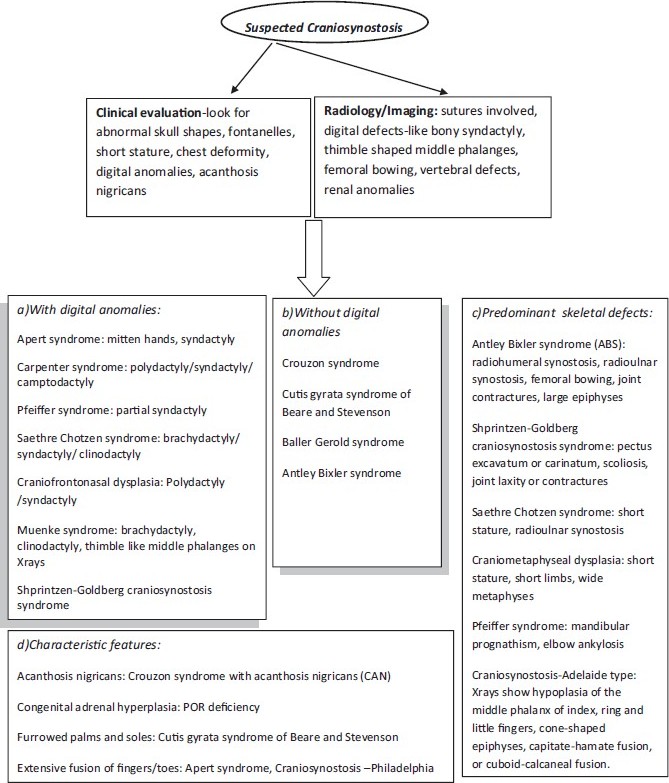 | Figure 3: Flow chart depicting approach to clinical diagnosis of craniosynostosis syndromes
Click here to view |
| References | | |
| 1. | Gleeson JG, Dobyns WB, Plawner L, Ashwal S. Congenital structural defects. In: Swaiman KF, Ashwal S, Ferriero DM, editors. Pediatric Neurology: Principles and Practice. 4 th ed. Philadelphia: Mosby, Elsevier; 2006. p. 441-54. 
|
| 2. | Online Mendelian Inheritance in Man. OMIM (TM). McKusick-Nathans Institute for Genetic Medicine, John Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD); 2008.
|
| 3. | Wilkie AO, Bochukova EG, Hansen RM, Taylor IB, Rannan Eliya SV, Byren JC, et al. Clinical dividends from the molecular genetic diagnosis of craniosynostosis. Am J Med Genet A 2007;143A:1941-9.
|
| 4. | Bonaventure J, El Ghouzzi V. Molecular and cellular bases of syndromic craniosynostoses. Expert Rev Mol Med 2003;5:1-17.
|
| 5. | Kapp-Simon KA, Speltz ML, Cunningham ML, Patel PK, Tomita T. Neurodevelopment of children with single suture craniosynostosis: A review. Childs Nerv Syst 2007;23:269-81.
|
| 6. | Jadico SK, Huebner A, McDonald-McGinn DM, Zackai EH, Young TL. Ocular phenotype correlations in patients with TWIST versus FGFR3 genetic mutations. J AAPOS 2006;10:435-44.
|
| 7. | Wilkes D, Rutland P, Pulleyn LJ, Reardon W, Moss C, Ellis JP, et al. A recurrent mutation, ala391glu, in the transmembrane domain of FGFR3 causes Crouzon syndrome and acanthosis nigricans. J Med Genet 1996;33:744-8.
|
| 8. | Arnaud-Lopez L, Fragaso R, Mantilla-Capacho J, Barros-Nunez P. Crouzon with acanthosis nigricans. Further delineation of the syndrome. Clin Genet 2007;72:405-10.
|
| 9. | Vogels A, Fryns JP. Pfeiffer syndrome. Orphanet J Rare Dis 2006;1:19.
|
| 10. | McGaughran J, Sinnott S, Susman R, Buckley MF, Elakis G, Cox T, et al. A case of Beare-Stevenson syndrome with a broad spectrum of features and a review of the FGFR2 Y375C mutation phenotype. Clin Dysmorphol 2006;15:89-93.
|
| 11. | Perlyn CA, Marsh JL. Craniofacial dysmorphology of Carpenter syndrome: Lessons from three affected siblings. Plast Reconstr Surg 2008;121:971-81.
|
| 12. | Doherty ES, Lacbawan F, Hadley DW, Brewer C, Zalewski C, Kim HJ, et al. Muenke syndrome (FGFR3-related craniosynostosis): Expansion of the phenotype and review of the literature. Am J Med Genet A 2007;143A:3204-15.
|
| 13. | Honnebier MB, Cabiling DS, Hetlinger M, McDonald-McGinn DM, Zackai EH, Bartlett SP. The natural history of patients treated for FGFR3-associated (Muenke-type) craniosynostosis. Plast Reconstr Surg 2008;121:919-31.
|
| 14. | Robinson PN, Neumann LM, Demuth S, Enders H, Jung U, König R, et al. Shprintzen-Goldberg syndrome: Fourteen new patients and a clinical analysis. Am J Med Genet A 2005;135:251-62.
|
| 15. | Cohen MM Jr. Jackson-Weiss syndrome. Am J Med Genet 2001;100:325-9.
|
| 16. | Miller WL, Huang N, Pandey AV, Fluck CE, Agarwal V. P450 oxidoreductase deficiency: A new disorder of steroidogenesis. Ann N Y Acad Sci 2005;1061:100-8.
|
| 17. | Miller WL. P450 oxidoreductase deficiency: A new disorder of steroidogenesis with multiple clinical manifestations. Trends Endocrinol Metab 2004;15:311-5.
|
| 18. | Orr DJ, Slaney S, Ashworth GJ, Poole MD. Craniofrontonasal dysplasia. Br J Plast Surg 1997;50:153-61.
|
| 19. | Tan TY, McGillivray G, Georgen SK, White SM. Prenatal magnetic resonance imaging in Gomez-Lopez-Hernandez syndrome and review of the literature. Am J Med Genet A 2005;138:369-73.
|
| 20. | Müller U, Steinberger D, Kunze S. Molecular genetics of craniosynostotic syndromes. Graefes Arch Clin Exp Opthalmol 1997;235:545-50.
|
| 21. | Cornejo-Roldan LR, Roessler E, Muenke M. Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome. Hum Genet 1999;104:425-31.
|
| 22. | Sood S, Eldadah ZA, Krause WL, Mc Intosh I, Dietz HC. Mutation in fibrillin-1 and the Marfanoid-craniosynostosis (Shprintzen-Goldberg) syndrome. Nat Genet 1996;12:209-11.
|
| 23. | Ma L, Golden S, Wu L, Maxson R. The molecular basis of Boston-type craniosynostosis: The Pro148->His mutation in the N-terminal arm of the MSX2 homeodomain stabilizes DNA binding without altering nucleotide sequence preferences. Hum Mol Genet 1996;5:1915-20.
|
| 24. | Twigg SR, Matsumoto K, Kidd AM, Goriely A, Taylor IB, Fisher RB, et al. The origin of EFNB1 mutations in craniofrontonasal syndrome: Frequent somatic mosaicism and explanation of the paucity of carrier males. Am J Hum Genet 2006;78:999-1010.
|
| 25. | Chinen Y, Kaname T, Yanagi K, Saito N, Naritomi K, Ohta T. Opitz trigonocephaly C syndrome in a boy with a de novo balanced reciprocal translocation t(3;18)(q13.13;q12.1). Am J Med Genet A 2006;140:1655-7.
|
| 26. | Eshel G, Rahat E, Reish O, Barr J. Neurodevelopmental and behavioral abnormalities associated with deletion of chromosome 9p. J Child Neurol 2002;17:50-1.
|
| 27. | Stratton RF, Dobbins WB, Greenberg F, DeSana JB, Moore C, Fidon G, et al. Interstitial deletion of (17)(p11.2p11.2): Report of six additional patients with a new chromosome deletion syndrome. Am J Med Genet 1986;24:421-32.
|
| 28. | Raybaud C, Di Rocco C. Brain malformation in syndromic craniosynostoses, a primary disorder of white matter: A review. Childs Nerv Syst 2007;23:1379-88.
|
| 29. | Kawamoto HK, Heller JB, Heller MM, Urrego A, Gabbay JS, Wasson KL, et al. Craniofrontonasal dysplasia: A surgical treatment algorithm. Plast Reconstr Surg 2007;120:1943-56.
|
| 30. | Clayman MA, Murad GJ, Steele MH, Seagle MB, Pincus DW. History of craniosynostosis surgery and the evolution of minimally invasive endoscopic techniques: The University of Florida experience. Ann Plast Surg 2007;58:285-7.
|
| 31. | Chang HP, Lin HC, Liu PH, Chang CH. Midfacial and mandibular morphometry of children with Class II and Class III malocclusions. J Oral Rehabil 2005;32:642-7.
|
| 32. | Kotrikova B, Krempien R, Freier K, Mühling J. Diagnostic imaging in the management of craniosynostoses. Eur Radiol 2007;17:1968-78.
|
[Figure 1], [Figure 2], [Figure 3]
[Table 1]
| This article has been cited by | | 1 |
Role of 99mTc-ECD SPECT in the Management of Children with Craniosynostosis |
|
| Mayadhar Barik,Minu Bajpai,Rashmi Ranajn Das,Arun Malhotra,Shasanka Shekhar Panda,Manas Kumar Sahoo,Sadanand Dwivedi | | BioMed Research International. 2014; 2014: 1 | | [Pubmed] | [DOI] | | | 2 |
A Case of Congenital Radioulnar Synostosis from Prehispanic Peru |
|
| A. R. Titelbaum,J. W. Verano | | International Journal of Osteoarchaeology. 2013; : n/a | | [Pubmed] | [DOI] | | | 3 |
Familial incidence and associated symptoms in a population of individuals with nonsyndromic craniosynostosis |
|
| Jaclyn Greenwood,Pamela Flodman,Kathryn Osann,Simeon A. Boyadjiev,Virginia Kimonis | | Genetics in Medicine. 2013; | | [Pubmed] | [DOI] | |
|
|
|
|