|
 
 |
|
ORIGINAL ARTICLE |
|
|
|
| Year : 2012 | Volume
: 18
| Issue : 1 | Page : 91-94 |
| |
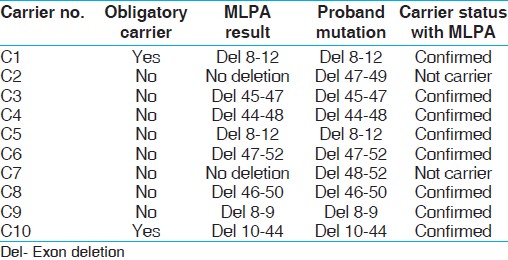
Utility of MLPA in mutation analysis and carrier detection for Duchenne muscular dystrophy
Prashant K Verma1, Ashwin Dalal2, Balraj Mittal1, Shubha R Phadke1
1 Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India
2 Diagnostics Division, Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India
| Date of Web Publication | 26-May-2012 |
Correspondence Address:
Shubha R Phadke
Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow - 226 014, Uttar Pradesh
India
 Source of Support: None, Conflict of Interest: None  | 3 |
DOI: 10.4103/0971-6866.96667

 Abstract Abstract | | |
Context: Multiplex ligation probe amplification (MLPA) is a new technique to identify deletions and duplications and can evaluate all 79 exons in dystrophin gene in patients with Duchenne muscular dystrophy (DMD). Being semi-quantitative, MLPA is also effective in detecting duplications and carrier testing of females; both of which cannot be done using multiplex PCR. It has found applications in diagnostics of many genetic disorders.
Aim: To study the utility of MLPA in diagnosis and carrier detection for DMD.
Materials and Methods: Mutation analysis and carrier detection was done by multiplex PCR and MLPA and the results were compared.
Results and Conclusions: We present data showing utility of MLPA in identifying mutations in cases with DMD/BMD. In the present study using MLPA, we identified mutations in additional 5.6% cases of DMD in whom multiplex PCR was not able to detect intragenic deletions. In addition, MLPA also correctly confirmed carrier status of two obligate carriers and revealed carrier status in 6 of 8 mothers of sporadic cases.
Keywords: Dystrophin, Duchenne and Becker muscular dystrophies, multiplex ligation-dependent probe amplification, carrier detection
How to cite this article:
Verma PK, Dalal A, Mittal B, Phadke SR. Utility of MLPA in mutation analysis and carrier detection for Duchenne muscular dystrophy. Indian J Hum Genet 2012;18:91-4 |
How to cite this URL:
Verma PK, Dalal A, Mittal B, Phadke SR. Utility of MLPA in mutation analysis and carrier detection for Duchenne muscular dystrophy. Indian J Hum Genet [serial online] 2012 [cited 2016 Jun 1];18:91-4. Available from: http://www.ijhg.com/text.asp?2012/18/1/91/96667 |
| Introduction | |  |
Duchenne muscular dystrophy (DMD; OMIM # 310200) is one of the most common genetic muscular dystrophies, with an estimated world-wide prevalence of approximately 1 in 3,500 males. [1] It is the most severe of the dystrophinopathies, and results from mutations in the DMD gene. The DMD gene consists of 79 exons, and spans ~2.4 Mb on short arm of X-chromosome. It has at least four promoters and is the largest known human gene. [2] The coding portion of the gene is however only about 14 kb long. It codes for the protein dystrophin, which is a membrane-associated protein present in muscle cells. Large deletions are the most common type of mutations in the DMD gene, accounting for up to ~65% of DMD cases, while duplications contribute to around 6-10% of the cases. Small insertions, deletions, point mutations and splicing defects, account for the remaining 25-30% of DMD cases. [3] Among these sub-exonic mutation sequences, nonsense mutations account for 34%, frameshifts for 33%, splice site mutations for 29%, and missense mutations for 4%. [4]
The multiplex PCR techniques described by Chamberlain [5] and Beggs [6] has been used extensively to detect deletions in DMD. However, the disadvantage with this method is that it only detects deletions in 20-30 out of 79 exons of DMD gene. The remaining deletions as well as any duplications cannot be detected by this method. Further, it cannot be used for carrier analysis in female relatives. In the past, Southern blotting, quantitative PCR, and multiplex amplifiable probe hybridization (MAPH) have been used to find out duplications in males and detection of female carriers, [7],[8],[9],[10] but these approaches has limited success in clinical practice. Recently, a new method, Mutiplex Ligation dependent Probe Amplification (MLPA), based on probe hybridization and amplification of target DNA, has been used for detection of deletions/duplications in DMD gene. [11] There are a number of studies, including two from India, emphasizing the utility of MLPA over other methods of mutation detection for DMD. [12],[13],[14],[15] However, there are very few studies worldwide, [16],[17] and none from India, regarding the utility of MLPA in carrier detection for female relatives of DMD patients. Here, we present the data regarding the utility of MLPA for both mutation detection in patients and carrier detection in families with DMD.
| Materials and Methods | | |
The study was conducted at a tertiary care hospital in India. The data of all patients referred between July 2009 and March 2011, with a clinical diagnosis of BMD or DMD and a raised level of creatinine phosphokinase, was analyzed. Carrier testing was done in female relatives after detection of mutation in the proband. All blood samples were obtained after taking informed consent from the patients/relatives.
Molecular analysis
DNA was isolated from leucocytes using standard salting-out protocols and quality and quantity of DNA was checked by nano-drop spectrophotometer. Multiplex PCR using twenty one primers belonging to proximal and distal hot-spot regions was carried out and patients who did not show any deletion by this method were considered for the study.
MLPA analysis was carried out according to the manufacturer's instructions (MRC Holland, Amsterdam, Netherlands). Briefly, 200-500 ng DNA was denatured and hybridized overnight at 60°C with the SALSA probe mix P034 (DMD exons 1-10, 21-30, 41-50 and 61-70) and P035 (DMD exons 11-20, 31-40, 51-60 and 71-79). Samples were then treated with DNA Ligase for 15 min at 54°C. The reaction was stopped by incubation at 98°C for 5 min. Finally, PCR amplification was carried out with the specific fluorescent labelled PCR primers. Amplification products were electrophoresed on an ABI PRISM 3100 Genetic Analyzer. The data obtained was analyzed by spreadsheet P034 and P035 provided by National Genetic Reference Laboratory Manchester. Patient result was considered as deleted if odd ratio was <1: 19 and duplicated if >19:1. Carrier result was considered as heterozygous deletion if dosage quotient was 0.35 to 0.65 and duplicated if dosage quotient was between 1.35 to 1.65 [Figure 1].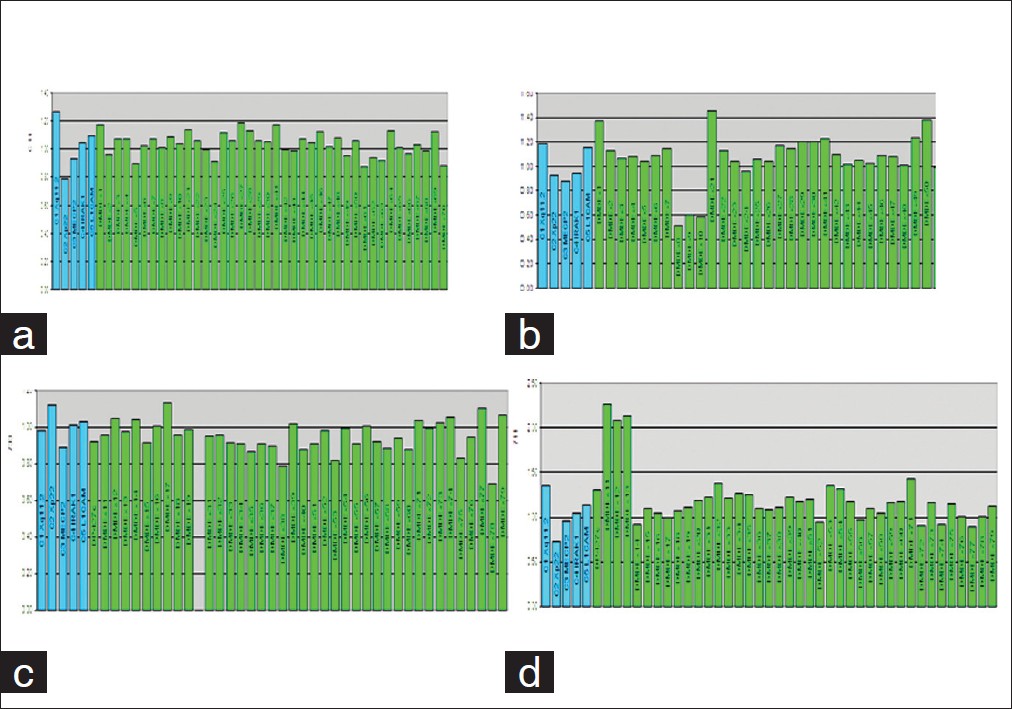 | Figure 1: (a) Normal MLPA peaks, (b) Heterozygous deletion in exons 8-10 in a carrier female, (c) Hemizygous deletion in exon 21, (d) Hemizygous duplication in exons 11-13
Click here to view |
| Results | | |
A total of 217 DMD cases were investigated for dystrophin gene mutation by multiplex PCR. Deletion mutations were identified in 161 (74.2%) cases by multiplex PCR and 56 cases (25.8%) did not show any deletion. Out of these 56 patients, five patients were not investigated further due to poor DNA quality. The remaining 51 cases were investigated by MLPA. MLPA helped in identification of deletions/duplications in 12 cases (23.5%) [Figure 1]. The deletions and duplications detected by MLPA were checked by reading-frame checker ver.1.9, available at the Leiden database (www.dmd.nl) and correlated clinically with patient phenotype. The details of mutations identified are shown in [Table 1]. Mutations were not identified in rest 39 cases (76.5%). These cases could probably account for point mutation or some other muscular dystrophy.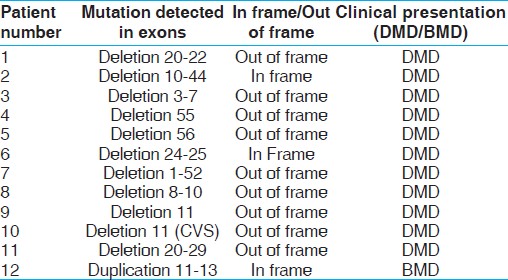 | Table 1: Mutations identified in patients by MLPA, which were not detected by multiplex PCR
Click here to view |
A total of ten females related to probands with known DMD mutation were included for carrier detection. Two mothers were obligate carriers and eight mothers were from families with sporadic cases. MLPA analysis detected mutation in 8 females (80%) including the two obligate carriers. The details are shown in [Table 2]. Two mothers did not show the mutation found in the respective probands, which could probably be due to occurrence of new mutation in the proband or gonadal mosaicism in the mother. Out of these, four mothers presented for antenatal testing by MLPA. Prenatal testing using MLPA detected one fetus to be affected and three to be normal for DMD.
| Discussion | | |
We have evaluated the clinical utility of MLPA for mutation detection, carrier identification, and antenatal diagnosis. We identified 11 patients with exonic deletions and one with exon duplication. Thus, use of MLPA resulted in identification of 12 additional cases (5.6%) with deletion/duplication, which were not detected by multiplex PCR. Most of the studies have reported detection of 5-8% additional cases by MLPA; hence, our results are in accordance with published literature. [14],[15],[18],[19]
Clinical phenotype depends upon the exact location of mutation in gene and not upon the size of deletion or duplication. An out-of-frame or frameshift deletion, irrespective of size, is expected to produce a severe phenotype, whereas an in-frame mutation is expected to result in a milder phenotype, namely Becker muscular dystrophy. Large in-frame deletions removing up to 35 exons in the central rod domain have been described for mild BMD. [20],[21],[22],[23] Reading-frame checker ver1.99 identified nine deletions as out-of-frame and three as in-frame deletions. [24] In one case, we found a large deletion of exon 1 to 52 resulting in DMD phenotype. Patient 2 and 6 had an in-frame deletion, but they showed DMD phenotypes. Reading-frame rule cannot be applicable for 9% cases as reported in Leiden database. There are 7% cases having in-frame deletion resulting in severe phenotype, whereas 2% having out-of-frame mutation, but resulting in BMD phenotype, as described in Leiden database. However, due to its inherent disadvantages, the reading frame rule cannot be used always to predict the phenotype.
Antenatal diagnosis using MLPA identified three fetuses to be unaffected and one to be affected. MLPA is the only method which can be used for prenatal diagnosis when the deletions/duplications are not detectable by multiplex PCR. Hence, we can provide accurate antenatal diagnosis and genetic counseling for 5.5% additional families by using MLPA for detection of mutation.
Carrier analysis is another major advantage of MLPA over conventional multiplex PCR. Out of the ten females analyzed for carrier status following detection of deletion/duplication in proband, eight females were detected as carriers for mutation by MLPA. Two females were obligate carriers. Six of eight mothers of sporadic cases of DMD were found to be carriers by MLPA. It has been observed that about two thirds of mothers of sporadic cases are carriers of mutation. Detection of carrier status is important for counselling the family regarding future pregnancies. Previously, Southern hybridization and semi-quantitative PCR based methods were used for carrier analysis but they are technically cumbersome and accuracy of results was also questionable. Now, MLPA is an easy method to detect carriers if mutation is known in the proband. Moreover, MLPA can also be used for determining positive carrier status even when the proband is deceased, although negative result cannot rule out carrier status.
Although no definitive therapy is available for DMD at this point of time, accurate detection of mutation is helpful in genetic counselling as well as prenatal diagnosis. The newer therapies like antisense oligonucleotide therapy are mutation specific and require the knowledge of mutation to select proper oligo for the patient. Therefore, MLPA can be used with confidence for identifying duplications and deletions in DMD patients as well as for carrier detection.
| References | | |
| 1. | Emery AE. Population frequencies of inherited neuromuscular diseases - a world survey. Neuromuscul Disord 1991;1:19-29. 
|
| 2. | Darras BT, Korf BR, Urion DK. Dystrophinopathies. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-1998 [updated 2008 March 21].
|
| 3. | Miller RG, Hoffman EP. Molecular diagnosis and modern management of Duchenne muscular dystrophy. Neurol Clin 1994;12:699-725.
|
| 4. | Chaturvedi LS, Mukherjee M, Srivastava S, Mittal RD, Mittal B. Point mutation and polymorphism in Duchenne/Becker muscular dystrophy (D/BMD) patients. Exp Mol Med 2001;33:251-6.
|
| 5. | Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 1988;16:11141-56.
|
| 6. | Beggs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet 1990;86:45-8.
|
| 7. | White S, Kalf M, Liu Q, Villerius M, Engelsma D, Kriek M, et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet 2002;71:365-74.
|
| 8. | Yau SC, Bobrow M, Mathew CG, Abbs SJ. Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J Med Genet 1996;33:550-8.
|
| 9. | Armour JA, Sismani C, Patsalis PC, Cross G. Measurement of locus copy number by hybridisation with amplifiable probes. Nucleic Acids Res 2000;28:605-9.
|
| 10. | White SJ, Sterrenburg E, van Ommen GJ, den Dunnen JT, Breuning MH. An alternative to FISH: Detecting deletion and duplication carriers within 24 hours. J Med Genet 2003;40:113.
|
| 11. | Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002;30:57.
|
| 12. | Janssen B, Hartmann C, Scholz V, Jauch A, Zschocke J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene:Potential and pitfalls. Neurogenetics 2005;6:29-35.
|
| 13. | Schwartz M, Duno M. Improved molecular diagnosis of dystrophin gene mutations using the multiplex ligation-dependent probe amplification method. Genet Test 2004;8:361-7.
|
| 14. | Kohli S, Saxena R, Thomas E, Singh J, Verma IC. Gene changes in Duchenne muscular dystrophy:Comparison of multiplex PCR and multiplex ligation-dependent probe amplification techniques. Neurol India 2010;58:852-6.
[PUBMED]  |
| 15. | Murugan S, Chandramohan A, Lakshmi BR. Use of multiplex ligation-dependent probe amplification (MLPA) for Duchenne muscular dystrophy (DMD) gene mutation analysis. Indian J Med Res 2010;132:303-11.
[PUBMED] |
| 16. | Pikó H, Vancsó V, Nagy B, Bán Z, Herczegfalvi A, Karcagi V. Dystrophin gene analysis in Hungarian Duchenne/Becker muscular dystrophy families - detection of carrier status in symptomatic and asymptomatic female relatives. Neuromuscul Disord 2009;19:108-12.
|
| 17. | Li H, Ding J, Wang W, Chen Y, Lu W, Shao H, et al. Combining approach with multiplex PCR and MLPA to detect deletion and duplication in DMD patients, carriers, and prenatal diagnosis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2009;26:318-22.
|
| 18. | White S, Kalf M, Liu Q, Villerius M, Engelsma D, Kriek M, et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet 2002;71:365-74.
|
| 19. | Pikó H, Vancsó V, Nagy B, Bán Z, Herczegfalvi A, Karcagi V. Dystrophin gene analysis in Hungarian Duchenne/Becker muscular dystrophy families - detection of carrier status in symptomatic and asymptomatic female relatives. Neuromuscul Disord 2009;19:108-12.
|
| 20. | Hoogerwaard EM, Bakker E, Ippel PF, Oosterwijk JC, Majoor- Krakauer DF, Leschot NJ, et al. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in The Netherlands: A cohort study. An unusual variant of Becker muscular dystrophy. Lancet 1999;353:2116-19.
|
| 21. | Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One 3. gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731-40.
|
| 22. | England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 1990;343:180-2.
|
| 23. | Mirabella M, Galluzzi G, Manfredi G, Bertini E, Ricci E, De Leo R, et al. Giant dystrophin deletion associated with congenital cataract and mild muscular dystrophy. Neurology 1998;51:592-5.
|
| 24. | Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006;34:135-44.
|
[Figure 1]
[Table 1], [Table 2]
| This article has been cited by | | 1 |
Genetic diagnosis of Duchenne and Becker muscular dystrophy using multiplex ligation-dependent probe amplification in Rwandan patients |
|
| A. Uwineza,J. Hitayezu,S. Murorunkwere,J. Ndinkabandi,C. K. Kalala Malu,J. H. Caberg,V. Dideberg,V. Bours,L. Mutesa | | Journal of Tropical Pediatrics. 2013; | | [Pubmed] | [DOI] | |
|
|
|
|