|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2012 | Volume
: 18
| Issue : 3 | Page : 363-365 |
| |
Renal amyloidosis due to familial Mediterranean fever misdiagnosed
Iman Hama1, Ratbi Ilham2, Naima Ouzeddoun3, Zaitouna Alhamany4, Radia Bayahia3, Abdelaziz Sefiani1
1 Human Genomic Center, Faculty of Medicine and Pharmacy, University Mohammed V Souissi, Rabat; Department of Medical Genomic, National Institute of Health, Rabat, Morocco
2 Department of Medical Genomic, National Institute of Health, Rabat, Morocco
3 Unit of Nephrology-Dialysis-Renal Transplantation, Ibn Sina University Hospital, Faculty of Medicine, Mohammed V University, Rabat, Morocco
4 Laboratory of Pathologic Anatomy, Children Hospital, Ibn Sina University Hospital, Rabat, Morocco
| Date of Web Publication | 4-Mar-2013 |
Correspondence Address:
Iman Hama
Department of Medical Genetics, National Institute of Health - 27, Avenue Ibn Batouta, B. P. 769 Rabat
Morocco
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.108043

 Abstract Abstract | | |
Familial Mediterranean fever (FMF, MIM 249100) is an autosomal recessive disease affecting mainly patients of the Mediterranean basin. It is an autoinflammatory periodic disorder characterized by recurrent episodes of fever and abdominal pain, synovitis, and pleuritis. The major complication of FMF is the development of renal AA amyloidosis. Treatment with colchicine prevents the occurrence of recurrent seizures and renal amyloidosis. The disease is caused by mutations in the MEFV gene. We report here the cases of two unrelated patients, who have been late diagnosed with FMF complicated by renal amyloidosis. We focus on the importance of early diagnosis of FMF, both to start rapidly treatment with colchicine and avoid renal amyloidosis, and to provide genetic counseling to families.
Keywords: Amyloidosis, diagnosis, familial mediterranean fever, MEFV gene, mutations
How to cite this article:
Hama I, Ilham R, Ouzeddoun N, Alhamany Z, Bayahia R, Sefiani A. Renal amyloidosis due to familial Mediterranean fever misdiagnosed. Indian J Hum Genet 2012;18:363-5 |
How to cite this URL:
Hama I, Ilham R, Ouzeddoun N, Alhamany Z, Bayahia R, Sefiani A. Renal amyloidosis due to familial Mediterranean fever misdiagnosed. Indian J Hum Genet [serial online] 2012 [cited 2016 Jun 1];18:363-5. Available from: http://www.ijhg.com/text.asp?2012/18/3/363/108043 |
| Introduction | |  |
Familial Mediterranean fever (FMF, MIM 249100) is an autosomal recessive disease characterized by recurrent episodes of fever accompanied with topical signs of inflammation mainly in the peritoneal, pleural, and articular cavities. [1] FMF is prevalent among populations living in the Mediterranean region. [2] Its prevalence ranges from 1/87 in Armenians [3] to 1/3500 in Turks. [4] The major complication of FMF is the development of renal AA amyloidosis. Treatment with colchicine prevents the occurrence of recurrent seizures and renal amyloidosis. [5] The disease is caused by mutations in the MEFV gene on 16p13, which is composed of 10 exons and encodes 781-amino-acid protein known as pyrin/marenostrin involved in the regulation of inflammation. [5],[6] To date, more than 150 gene alterations (mutations/polymorphisms) located in the MEFV gene have been identified. [7] The M694V, V726A, M694I, M680I mutations in exon 10 are the most frequent among all populations, and are associated with the most severe clinical pictures with the risk of AA amyloidosis. [2]
In Morocco, the disease remains under or late diagnosed, at the stage of renal complications. We report on two patients, who were not on colchicine prophylaxis, and developed chronic renal failure because their FMF statute was not known before they developed renal amyloidosis.
| Case Reports | | |
Case 1
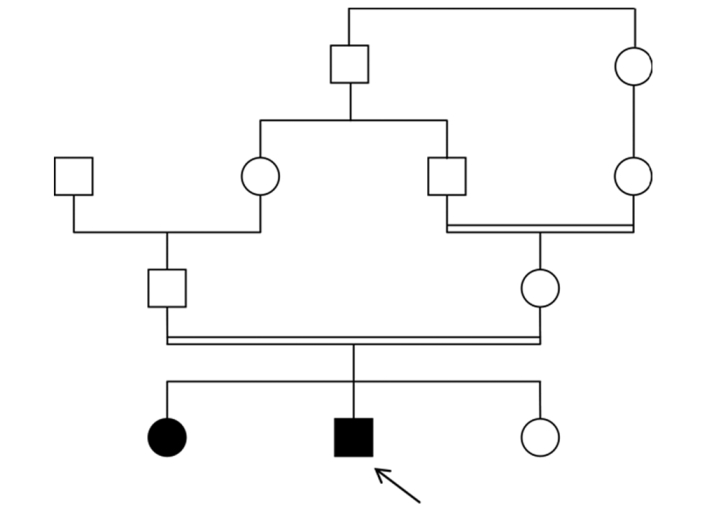
A 16-year-old boy was referred for end-stage renal failure caused by renal amyloidosis of the AA type. He had been subject, for 13 years, to recurrent episodes of fever lasting two to three days and accompanied by abdominal and joint pains. He had a partial response to nonsteroidal antiinflammatory drugs and corticosteroids. He is consanguineous in the first degree, the third of four children, with a positive family history of recurrent episodes of fever, less severe in his older sister of 18 years old [Figure 1]. The diagnosis of FMF was considered. The proband's parents gave informed consent for the genetic studies reported here. Peripheral blood was collected from the proband, his sister and their parents. Molecular genetic testing for FMF was performed by DNA sequencing of exon 10 of the MEFV gene, as previously reported. As a result, M694V mutation in homozygosis was determined in both patients. As expected the parents were heterozygote carriers of M694V mutation. Molecular analysis of his younger sister allowed us to comfort her about her status of normal homozygote after an episode of articular pain that alarm her parents because of the familial history of FMF.
Case 2
The patient is a 30-year-old female. She has been having periodic episodes of high fever, usually lasting less than 3 days, at 1-to 3-month intervals since she was 22 years old. The fever was often accompanied by diarrhea and abdominal pain. The patient had no medical follow-up for several months. During a severe episode, she was admitted to hospital and diagnosed with AA amyloidosis responsible of a chronic renal failure. The patient has no family history of recurrent episodes of fever. Her parents are not consanguineous. The symptomatic occurrence of periodic fevers with spontaneous improvement within 3 days suggested a possible diagnosis of FMF. After obtaining informed consent, DNA sequencing of exon 10 of the MEFV gene was performed. We found that the patient is compound heterozygous for V726A and A744S mutations.
| Discussion | | |
As a Mediterranean population, Maghrebians are particularly prone to develop FMF. The Department of Medical Genetics at the National Institute of Health in Rabat, considered a major medical genetics pole in Morocco, has taken interest in this pathology for a decade. This interest is motivated by two elements: The relatively high frequency of the disease among North African populations, which is estimated at 1% [8] and the high level of consanguineous marriages in our society (15.25%) that leads to a high incidence of autosomal recessive disorders. [9] We performed molecular diagnosis of FMF by screening for mutations of exons 10 and 2, as previously described. [5],[10],[11] We also worked on the characterization of the mutational profile of this disease among the Moroccan population. As such, we have recently reported data on MEFV gene mutations causing FMF among Moroccans patients. [12] The most frequent mutation in Moroccan patients is M694V (47 %), followed by M694I (32 %), A744S (6.5 %), M680L (4 %), M694del (2%), and E148Q (6.5 %). There are no pathognomonic clinical symptoms of FMF and any specific biochemical abnormality. A positive response to colchicine treatment makes a good case for the disease, but only genetic studies are considered a clinical proof of the disease. [13] Early diagnosis of FMF is crucial to avoid year-long roundtrip visits to the hospital to get a diagnosis and even sometimes undergo unnecessary abdominal surgery because of the symptom of abdominal pain. The early and accurate diagnosis of FMF is important all the more so as colchicine has proved to be an effective and cheap treatment, helping to reduce significantly the frequency and duration of attacks and to delay renal complication. [14] In spite of all these clinical and epidemiological data, FMF remains a little-known and largely underestimated pathology in Morocco's daily medical practice, which does not allow patients to get a diagnosis in specialized centers. The two clinical cases reported highlight the importance of an early diagnosis of FMF to introduce the appropriate therapeutic to prevent renal amylodoisis and for genetic counseling of families. The first case reported here is homozygous for the M694V, a dominant mutation in our population associated with a severe phenotype. [15] His sister, who has the same genotype, has a less severe clinical picture. This intrafamilial variability in the clinical expression can be explained by the action of modifying genes in the MEFV gene. The second case is compound heterozygous mutations of V726A and A744S. The A744S mutation is common in our population. [10] The V726A mutation is the second most frequent mutation in Arabs from Middle East, in Armenians and Turks, [16] while it was nonexistent in our population. [12] The patient's ancestors are Moroccan going back three generations. We assume that she might have an earlier nonMaghrebian ancestor. The data reported by our preliminary study, [12] and by the two cases, are prone to bring Moroccan clinicians' attention to the relative frequency of this disease in Morocco. We encourage them to consider this pathology in their daily practice and to work in concert with geneticians and epidemiologists to run wider studies to characterize the clinical and molecular profile of FMF in our country and contribute to take better care of our patients. Moreover, the present study proves the importance of early diagnosis of FMF, both to start rapidly treatment with colchicine and avoid renal amyloidosis, and to provide genetic counseling to families.
| References | | |
| 1. | Ben-Chetrit E, Levy M. Familial Mediterranean fever. Lancet 1998;351:659-64. 
[PUBMED] |
| 2. | Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum 2009;61:1447-53.
[PUBMED] |
| 3. | Sarkisian T, Ajrapetian H, Beglarian A, Shahsuvarian G, Egiazarian A. Familial Mediterranean fever in Armenian population. Georgian Med News 2008;156:105-11.
[PUBMED] |
| 4. | Tunca M, Akar S, Onen F, Ozdogan H, Kasapcopur O, Yalcinkaya F, et al. For the Turkish FMF Study Group. Familial Mediterranean fever (FMF) in Turkey: Results of a nationwide multicenter study. Medicine (Baltimore) 2005;84:1-11.
[PUBMED] |
| 5. | International FMF Consortium. Ancient missense mutations in a new member of the Ro- Ret gene family are likely to cause familial mediterranean fever. Cell 1997;90:787-807.
|
| 6. | The French FMF Consortium. A candidate gene for familial Mediterraneand fever. Nat Genet 1997;17:25-31.
|
| 7. | Available from: http://www.fmf.igh.cnrs.fr/ISSAID/infevers/ [Last accessed on 2012 Dec 23].
|
| 8. | Belmahi L, Sefiani A, Fouveau C, Feingold J, Delpech M, Grateau G, et al. Prevalence and distribution of MEFV mutations among Arabs from the Maghreb patients suffering from familial Mediterranean fever. C R Biol 2006;329:71-4.
[PUBMED] |
| 9. | Jaouad IC, Elalaoui SC, Sbiti A, Sbiti A, Elkerh F, Belmahi L, et al. Consanguineous marriages in Morocco and the consequence for the incidence of autosomal recessive disorders. J Biosoc Sci 2009;41:575-81.
[PUBMED] |
| 10. | Bernot A, da Silva C, Petit JL, Cruaud C, Caloustian C, Castet V, et al. Non-founder mutations in the MEFV gene establish his gene as the cause of familial Mediterranean fever (FMF). Hum Mol Genet 1998;7:1317-25.
[PUBMED] |
| 11. | Eisenberg S, Aksentijevich I, Deng Z, Kastner DL, Matzner Y. Diagnosis of familial Mediterranean fever by a molecular genetics method. Ann Intern Med 1998;129:539-42.
[PUBMED] |
| 12. | Belmahi L, Cherkaoui IJ, Hama I, Sefiani A. MEFV mutations in Moroccan patients suffering from familial Mediterranean Fever. Rheumatol Int 2011;32:981-4.
[PUBMED] |
| 13. | Livneh A, Langevitz P. Diagnostic and treatment concerns in familial Mediterranean fever. Baillieres Best Pract Res Clin Rheumatol 2000;14:477-98.
[PUBMED] |
| 14. | Goldfinger SE. Colchicine for familial Mediterranean fever [letter]. N Engl J Med 1972;287:130.
[PUBMED] |
| 15. | Majeed HA, El-Shanti H, Al-Khateeb MS, Rabaiha ZA. Genotype/phenotype correlations in Arab patients with familial Mediterranean fever. Semin Arthritis Rheum 2002;31:371-6.
[PUBMED] |
| 16. | Majeed HA, El-Khateeb M, El-Shanti H, Rabaiha ZA, Tayeh M, Najib D. The Spectrum of Familial Mediterranean fever Gene Mutations in Arabs: Report of a Large Series. Semin Arthritis Rheum 2005;34:813-8.
[PUBMED] |
[Figure 1]
|