|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2012 | Volume
: 18
| Issue : 3 | Page : 376-378 |
| |
Orofaciodigital syndrome type-VI (Varadi-Papp syndrome) with several Y-shaped metacarpals
Pulak R Mahato1, Shashi B Pandey2
1 Department of Paediatrics, Paediatric Unit, S. D. Hospital, Jhargram, India
2 Department of Radio-Diagnosis, Radiology Unit, Spandan Hospital, Medinipur, West Bengal, India
| Date of Web Publication | 4-Mar-2013 |
Correspondence Address:
Pulak R Mahato
Senior Medical Officer (Paediatrician), Sub-Divisional Hospital, Jhargram, Dist. Paschim Medinipur, West Bengal - 721 507
India
 Source of Support: None, Conflict of Interest: None  | 1 |
DOI: 10.4103/0971-6866.108053

 Abstract Abstract | | |
Orofaciodigital syndrome type-VI (Varadi-Papp Syndrome) is a rare autosomal recessive disorder characterized by variable orofacial anomalies, central polydactyly of the hands, and cerebellar dysgenesis (mainly hypoplasia or aplasia of vermis, rarely Dandy-Waker anomaly). Here a case of Varadi-Papp syndrome with recurrent episodic tachypnea-apnea, minimal orofacial features, several Y-shaped metacarpals, and cerebellar vermis hypoplasia, diagnosed in the neonatal age, is reported for the first time in Indian literature. The importance of early accurate diagnosis of this rare disease for proper genetic counseling and prenatal case detection of pregnancy at risk is also emphasized as the prognosis is poor in almost all cases.
Keywords: Indian, neonatal, orofaciodigital syndrome type-VI, Varadi-Papp syndrome
How to cite this article:
Mahato PR, Pandey SB. Orofaciodigital syndrome type-VI (Varadi-Papp syndrome) with several Y-shaped metacarpals. Indian J Hum Genet 2012;18:376-8 |
How to cite this URL:
Mahato PR, Pandey SB. Orofaciodigital syndrome type-VI (Varadi-Papp syndrome) with several Y-shaped metacarpals. Indian J Hum Genet [serial online] 2012 [cited 2016 Jun 1];18:376-8. Available from: http://www.ijhg.com/text.asp?2012/18/3/376/108053 |
| Introduction | |  |
Orofaciodigital syndrome type-VI (OFDS-VI) is a very rare autosomal recessive disorder distinguished from other types of OFDS by cerebellar dysgenesis and manual central polydactyly with y-shaped central metacarpals. [1] The syndrome was first reported by Varadi, et al.[2] in six children in an endogamic gypsy colony of Hungary. OFDS-VI is currently classified as one of the Joubert syndrome-related disorders [3] because of similar clinical presentation and presence of the molar tooth sign (cerebellar vermis hypoplasia and brainstem anomalies) on neuroimaging. Here, we report, for the first time in India, a case of OFDS-VI (Varadi-Papp syndrome) with some additional merits like early diagnosis in the neonatal age and presence of several y-shaped meta-carpals.
| Case Report | | |
A 17-day-old girl was admitted with chief complaint of episodic respiratory distress noted since the second day of her life. The baby was born out of nonconsanguinous marriage, by normal delivery at term, to a primigravida mother with an uneventful pregnancy, and cried immediately after birth. There was no maternal history of teratogenic drug or radiation exposure. No antenatal ultrasonography was done. After her birth (weight 2000 g) in a rural hospital, congenital malformations were first noted. The family history was negative for congenital anomaly and mental retardation. On admission her weight was 1750 g (SGA), length 42 cm (<5 th centile), OFC 32 cm (<5 th centile), and respiratory rate was 42/min with episodic panting type of respiration, with rate up to 100/min lasting for 1-3 minutes, followed by apnea for 12-15 s without bradycardia or cyanosis. In addition, physical examinations revealed posteriorly rotated low set ears, prominent upper forehead, sublingual nodules, bimanual heptadactyly, bilateral hallucal polysyndactyly, and an accessory right postaxial toe, coarse crepitations on the right chest, and generalized hypotonia with diminished deep tendon jerks. Neonatal reflexes were depressed. The other findings were unremarkable and fundoscopic examination was normal. The following investigations were done and found to be normal-routine blood and urine, serum CRP, blood sugar, blood urea, SGPT, serum calcium, X-rays of pelvis and skull, abdominal ultrasonography, and echocardiography [Figure 1] and [Figure 2].Radiography of chest revealed pneumonitic changes in the right mid- and upper zones. X-ray of the hands showed y-shaped right third, left fourth and left fifth metacarpals with bimanual central polydactyly, a right postminimus, and a left postaxial extra digit. In addition, X-ray revealed short middle phalanx of all fingers. X-rays of the feet showed duplication of the first metatarsal and hallucal phalanges, an extra epiphysis in the soft-tissue hallux between normal and accessory bony hallux and syndactyly of first three toes, bilaterally. MRI of brain demonstrated a hypoplastic cerebellar vermis with a molar tooth sign (which consists of deepened interpeduncular fossa, thickened superior cerebellar peduncles and hypoplasia of cerebellar vermis) on axial image at the level of the isthmus. The diagnosis was reached based on clinicoradiological findings. She was treated conservatively and episodic tachypnea-apnea gradually decreased at 1 month of age. The child was then discharged and advised to seek regular follow-up for further evaluation and possible supportive interventions. Although advised, VEP and BAER test were not carried out. A chromosomal study (conventional G-banding) showed a normal karyotype (46, XX). At 2½ months of age, examination revealed growth failure (weight 2500 g), feeding difficulty, hypotonia, absent social smile, poor visual fixation and occasional episodic tachypnea.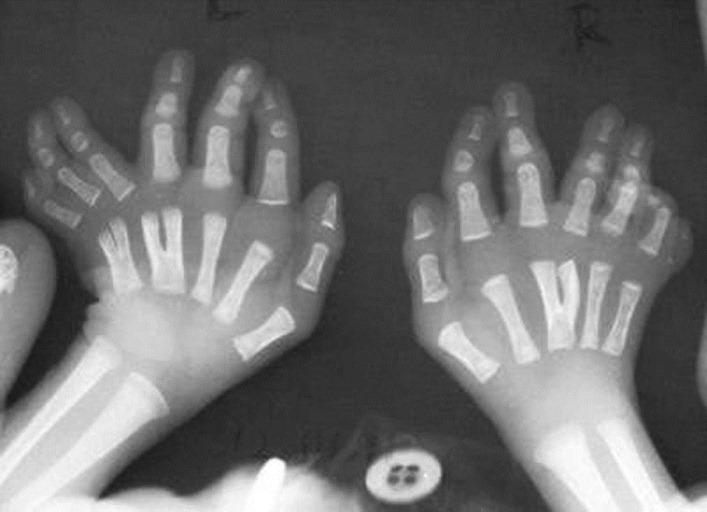 | Figure 1: X rays of the hands showing Y-shaped right third, left fourth, and left fifth metacarpals
Click here to view |
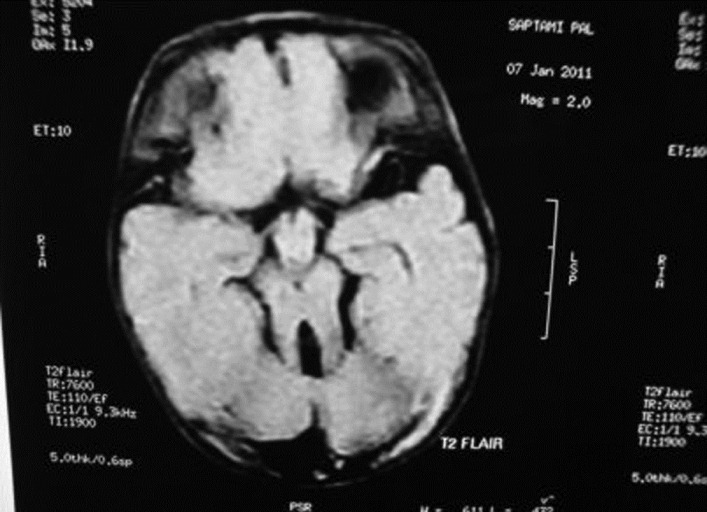 | Figure 2: MRI-brain (T2 FLAIR) showing cerebellar vermis hypoplasia with the "molar tooth sign"
Click here to view |
| Discussion | | |
In 1980, Varadi, et al. reported a syndrome of polydactyly, cleft-lip/palate, lingual lump, somatic and psychomotor retardation with autopsy evidence of absent cerebellar vermis in the proband. The inheritance pattern was suggested to be autosomal recessive. Muenke, et al., [4] after adding three similar patients, first reviewed and delineated clinical features of OFDS-VI. The authors pointed out that cerebellar anomalies (vermis hypoplasia/aplasia or Dandy-Walker variants) are consistent findings in Varadi-Papp syndrome, in addition to variable orofacial features and polysyndactyly. Manual central polydactyly, defined as a fully formed extra digit arising from an additional central metacarpal or from a bifid/Y-shaped, usually, third metacarpal, was considered as the most frequent and specific diagnostic feature. In addition to central polydactyly, both postaxial [2],[4],[5] and preaxial [4],[6] manual polydactyly are common, as are clinodactyly and syndactyly. [4],[5] The feet usually show preaxial-polydactyly [2],[4],[5],[6] without [6] or with cutaneous syndactyly. [4],[5] Oral findings commonly include lingual or sublingual-lump/nodules, [2],[4],[5],[7] oral frenula, [4] and highly arched and/or cleft-palate. [2],[4],[5],[6] Common facial features are cleft-lip, [2],[4],[6] broad nasal tip/bridge, [2],[4],[5] hypertelorism, [2],[4],[6] epicanthic folds, [2],[4],[5] abnormal eye movements, [4],[5],[7] strabismus, [2],[4] and low set ears. [2],[6] Psychomotor retardation is almost universal with rare exception. [4] Episodic tachypnea, [4],[7] hypotonia and/or ataxia, [2],[4],[5],[7] growth failure, [2],[4] and deafness [4] are often reported. Hypogonadism and/or cryptorchidism with micropenis, [2],[4] hypoplastic epiglottis, [6] aortic stenosis, [2] hypothalamic hamartoma, [7] and subcortical-heterotopias [7] are also noted. Our patient presented with all features consistent with the diagnosis of OFDS-VI. However, orofacial features were minimal with the absence of cleft-lip/palate and abnormal ocular movements whereas the metacarpal findings were remarkable. The presence of several y-shaped metacarpals with a unique combination of involvement of third metacarpal on one hand and fourth and fifth metacarpi on the other was the remarkable feature of the patient. Because of phenotypic overlap, Pallister-Hall syndrome (PHS) is considered as one of differential diagnoses. [1],[4] Clinico-radiological features that distinguish the syndromes are bifid epiglottis, imperforate anus, insertional-polydactyly and lethal airway-anomalies in PHS, and manual central polydactyly with cerebellar vermis hypoplasia in OFDS-VI. Moreover, by a molecular cytogenetic test, mutations in the GLI3 gene are detected in approximately 95% patients with PHS. [8] Although most of the previously described patients with OFDS-VI showed normal chromosomal study, recently, mutations in the TMEM216 gene have been identified in two cases. [9] Oral frenula, lingual/sublingual nodule, and central polydactyly distinguish Varadi-Papp syndrome from other subgroups of Joubert syndrome-related disorders.
Despite the fact that Varadi-Papp syndrome is very rare in India, an early accurate diagnosis is necessary for proper genetic counseling and prognostication. Treatments are mainly symptomatic and supportive with reconstructive surgery for correctable malformations. Prenatal diagnosis of the pregnancy at risk is possible [4],[10] and advisable, and termination of pregnancy is indicated [11] as the prognosis is almost always poor - either early death or survival with psychomotor retardation.
| Conclusion | | |
Orofaciodigital syndrome type-VI, a rare genetic disease with poor prognosis, should always be considered as a differential diagnosis in neonates presenting with episodic tachypnea especially if poly (syn) dactyly is present and meticulous orofacial examination should be done. Evidence of central polydactyly of the hands and cerebellar anomalies in the presence of minimal orofacial manifestation establishes the diagnosis of the disorder. Management includes genetic counseling and possible medicosurgical supportive interventions.
| Acknowledgments | | |
We thank Dr. S. Kanrar (Superintendent, S. D. Hospital, Jhargram, Paschim Medinipur, W.B.), for granting us permission to publish this case report.
| References | | |
| 1. | Gurrieri F, Franco B, Toriello H, Neri G. Oral-Facial-Digital Syndromes: Review and Diagnostic Guidelines. Am J Med Genet A 2007;143A: 3314-23. 
[PUBMED] |
| 2. | Váradi V, Szabó L, Papp Z. Syndrome of polydactyly, cleft lip/palate or lingual lump and psychomotor retardation in endogamic gypsies. J Med Genet 1980;17:119-22.
|
| 3. | Parisi M A, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders). Eur J Hum Genet 2007;15:511-21.
|
| 4. | Münke M, McDonald DM, Cronister A, Stewart JM, Gorlin RJ, Zackai EH. oral-facial-digital syndrome Type VI (V'aradi syndrome): Further clinical delineation. Am J Med Genet 1990;35:360-9.
|
| 5. | Mauceri L, Greco F, Baieli S, Sorge G. Varadi-Papp syndrome: Report of a case. clin dysmorphol 2000;9:289-90.
|
| 6. | Hayes LL, Simoneaux SF, Palasis S, Niyazov DM. Laryngeal and tracheal anomalies in an infant with oral-facial-digital syndrome type VI (Váradi-Papp): Report of a transitional type. Pediatr Radiol 2008;38:994-8.
|
| 7. | Takanashi J, Tada H, Ozaki H, Barkovich AJ. Malformations of cerebral cortical development in oral-facial-digital syndrome Type VI. AJNR Am J Neuroradiol 2009;30:e22-3.
|
| 8. | Johnston JJ, Olivos-Glander I, Killoran C, Elson E, Turner JT, Peters KF, et al. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: Robust phenotype prediction from the type and position of GLI3 mutations. Am J Hum Genet 2005;76:609-22.
|
| 9. | Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes . Nat Genet 2010;42:619-25.
|
| 10. | Poretti A, Brehmer U, Scheer I, Bernet V, Boltshauser E. Prenatal and neonatal MR imaging findings in oral-facial-digital syndrome type VI. AJNR Am J Neuroradiol 2008;29:1090-1
|
| 11. | Varadi V, Papp Z. 25 years' history of Varadi-Papp syndrome (orofaciodigital syndrome VI). Orv Hetil 2005;146:2017-22.
|
[Figure 1], [Figure 2]
| This article has been cited by | | 1 |
Oral-facial-digital syndrome with mesoaxial polysyndactyly, common AV canal, hirschsprung disease and sacral dysgenesis: Probably a transitional type between II, VI, variant of type VI or a new type |
|
| Rabah M. Shawky,Heba Salah Abd Elkhalek,Marwa M. Al-Fahham,Shaimaa Abdelsattar Mohammad,Shaimaa Gad | | Egyptian Journal of Medical Human Genetics. 2014; | | [Pubmed] | [DOI] | |
|
|
|
|









