|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 1 | Page : 104-107 |
| |
MICrocephaly, disproportionate pontine and cerebellar hypoplasia syndrome: A clinico-radiologic phenotype linked to calcium/calmodulin-dependent serine protein kinase gene mutation
Rashid Saleem, Gururaj Setty, Nahin Hussain
Department of Pediatric Neurology, Leicester Royal Infirmary, University Hospitals of Leicester NHS Trust, United Kingdom
| Date of Web Publication | 4-Jun-2013 |
Correspondence Address:
Rashid Saleem
Department of Pediatric Neurology, Leicester Royal Infirmary, University Hospitals of Leicester NHS Trust, Leicester, LE1 5WW
United Kingdom
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.112921

 Abstract Abstract | | |
MICrocephaly, disproportionate pontine and cerebellar hypoplasia (MICPCH) syndrome, a rare X-linked disorder, generally seen in girls, is characterized by neurodevelopmental delay, microcephaly, and disproportionate pontine and cerebellar hypoplasia. It is caused by inactivating calcium/calmodulin-dependent serine protein kinase (CASK) gene mutations. We report a 2-year-old girl with severe neurodevelopmental delay, microcephaly, minimal pontine hypoplasia, cerebellar hypoplasia, and normal looking corpus callosum, with whom the conventional cytogenetic studies turned out to be normal, and an array-comparative genomic hybridization (a-CGH) analysis showed CASK gene duplication at Xp11.4. Our case highlights the importance of using clinico-radiologic phenotype to guide genetic investigation and it also confirms the role of a-CGH analysis in establishing the genetic diagnosis of MICPCH syndrome, when conventional cytogenetic studies are inconclusive.
Keywords: Array-comparative genomic hybridization, calcium/calmodulin-dependent serine protein kinase gene, microcephaly, X-chromosome inactivation, X-linked neurodevelopmental delay
How to cite this article:
Saleem R, Setty G, Hussain N. MICrocephaly, disproportionate pontine and cerebellar hypoplasia syndrome: A clinico-radiologic phenotype linked to calcium/calmodulin-dependent serine protein kinase gene mutation. Indian J Hum Genet 2013;19:104-7 |
How to cite this URL:
Saleem R, Setty G, Hussain N. MICrocephaly, disproportionate pontine and cerebellar hypoplasia syndrome: A clinico-radiologic phenotype linked to calcium/calmodulin-dependent serine protein kinase gene mutation. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:104-7. Available from: http://www.ijhg.com/text.asp?2013/19/1/104/112921 |
| Introduction | |  |
Array-comparative genomic hybridization (a-CGH) assay has made the genome-wide screening of genomic copy-number variations (CNVs) possible, enabling to assign genetic diagnoses to phenotypes, which could not be accounted for in the past. [1] MICrocephaly, disproportionate pontine and cerebellar hypoplasia (MICPCH) syndrome [Online Mendelian Inheritance in Man (OMIM) number 300749], a rare clinico-radiological phenotype, was first described by Najm et al. and linked to calcium/calmodulin-dependent serine protein kinase (CASK) gene mutations. [2],[3] The core phenotype comprises severe neurodevelopmental delay, microcephaly, and disproportionate pontine and cerebellar hypoplasia. Moog [4] has further defined the phenotypical spectrum of the syndrome. Neuroradiological features associated with CASK mutations have been refined by Takanashi. [5] To date, forty five or so female patients with characteristic phenotype and two male patients with lethal form have been reported. [2],[4],[6],[7],[8] We report a girl with clinico-radiologic features of MICPCH syndrome, whose conventional cytogenetic analysis was normal and a subsequent a-CGH analysis discovered microduplication in CASK gene at Xp11.4.
| Case Report | | |
A 2-year-old Asian girl was referred to pediatric neurology service with global developmental delay and growth restriction. She could sit unaided and roll over, but was unable to stand without support. She could not speak clear meaningful words.
She was born at term by normal vaginal delivery to un-related healthy parents. Pregnancy was unremarkable for infection, exposure to known teratogens, polyhydramnios or decreased fetal movements. She did not require resuscitation at birth or admission to neonatal unit. Her head circumference at birth was below 0.4 th centile. She was both breast and bottle fed. During the weaning period, she struggled with lumpy food and failed to gain weight beyond 6 months. Consequently, she was started on nasogastric tube feeding. Family history was unremarkable for developmental delay or microcephaly.
Clinical assessment revealed microcephaly, low weight (<0.4 th centile), an open mouthed appearance with thickened alveolar ridges, prominent maxilla, bilateral epicanthic folds, short nose, long philtrum, large ears, hypoplastic 5 th toe nails, delayed neurodevelopmental milestones, low axial tone, and slightly increased peripheral tone, more so in the lower limbs. Her audiological and ophthalmological assessments were unremarkable. Physiotherapy and Speech and Language Therapy, and dietetic inputs were in place.
As a part of her diagnostic work-up, she underwent extensive investigations. Magnetic resonance imaging (MRI) showed prominent lateral ventricles, mega cisterna magna, small cerebellum, and normal corpus callosum [Figure 1]. Neurometabolic investigations were normal and Toxoplasmosis, Rubella, Cytomegalovirus, Herpes simplex (TORCH) studies were negative.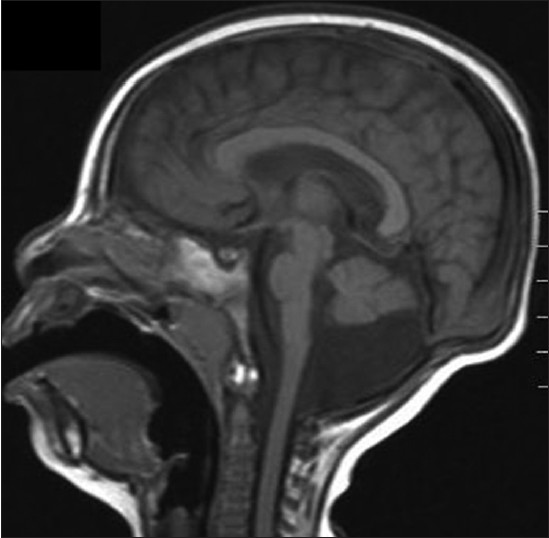 | Figure 1: T1 - weighted sagittal image of a 2 - year - old girl. Note the hypoplastic pons and cerebellum with normal appearance of the corpus callosum
Click here to view |
She was referred to clinical genetics team with a view to assigning a syndromic diagnosis. Conventional cytogenetic studies were reported normal. Subsequently, an a-CGH analysis was carried out, which revealed CASK gene duplication at Xp11.4.
| Discussion | | |
MICPCH syndrome is a rare X-linked genetic disorder caused by CASK gene mutations and is characterized by neurodevelopmental delay and specific brain malformations. [2],[3] CASK, a multi-domain scaffolding protein, is a member of membrane associated guanylate kinase protein family, [9] which regulates presynaptic neurotransmitter release, postsynaptic ion-channel function, and expression of genes associated with brain development. [10],[11] Evidence suggests that loss-of-function CASK gene mutations lead to X chromosome inactivation, causing severe phenotype, whereas hypomorphic missense CASK mutations produce less severe phenotypes. [4] Deletions and intragenic mutations have been associated with MICPCH syndrome more commonly than duplications. [3],[4] Inversion of the duplicated fragment results in a null allele. [4] Almost invariably, the CASK genetic aberrations are de novo, resulting most commonly from the mutational mechanism of non-allelic recombination. [2]
Najm [2] studied four females and one male who died in neonatal period to describe the core phenotype and found CNVs, gene inversion and single nucleotide polymorphism as the possible mutational mechanisms. Hayashi [7] in his ten cases discovered nonsense mutations, 2-bp deletions, exon-intron junction mutations, interstitial duplications, and heterozygous deletions with almost equal frequency. Moog [4] reported 20 patients of which 11 showed submicroscopic CNVs, including nine deletions and two duplications in the CASK gene. Recently, Burglen [8] have reported 13 patients (11 females and 2 males) with de novo CASK gene mutations, 10 being intragenic mutations and 3 Xp11.4 deletions.
The core MICPCH syndrome clinical phenotype, originally described by Najm, [2] was further defined by Moog, [4] and confirmed by Burglen. [8] It includes severe neurodevelopmental delay, varying degrees of congenital microcephaly, severe postnatal microcephaly (occipitofrontal circumference <3.5 SD) by 1 year of age, commonly associated with feeding difficulties, postnatal growth restriction, axial hypotonia, peripheral hypertonia, and occasional optic nerve hypoplasia and/or other eye abnormalities and sensorineural hearing impairment. Dysmorphic features comprised prominent and/or broad nasal bridge and tip, small or short nose, long philtrum, small chin, and/or large ears, hypoplastic toe nails etc. [4]
Takanashi [5] described the radiological phenotypic spectrum associated with CASK gene mutations, which confirmed the findings of Najm [2] and was, in turn, corroborated by observations recorded by Moog [4] MRI revealed varying degrees of brainstem and cerebellar hypoplasia and in some cases, showed increase in the fourth ventricle size, subtle changes in gyral pattern in frontal cortex, and mild dilatation of lateral ventricles. [2],[4],[5] Takanashi [5] also concluded that a relatively normal corpus callosum with low cerebrum/corpus callosum ratio in a girl with microcephaly and neurodevelopmental delay, alludes to the possibility of CASK gene mutation, as the pontocerebeller hypoplasia in non-CASK gene mutations is associated with thinning of corpus callosum.
Our case fits in the phenotypic and genotypic features of MICPCH syndrome described above. Severe neurodevelopmental delay, marked microcephaly, small cerebellum, prominent lateral ventricles, and dysmorphic features such as short nose, long philtrum, prominent maxilla, and large ears fit in the clinico-radiologic phenotype described. [2],[4],[5],[8] Normal appearing corpus callosum (CC) in our patient is in line with observations made by Takanashi. [5] Minimal vermis involvement in our case was reported by Moog [4] in patient 10, 11, and 12. As seen in our case, hypoplastic toe nails were documented in patient 7 by Moog [4] and patient 10 by Burglen. [8] Fourth ventricle size was near normal in our case as was reported by Moog [4] in five of their patients. Although, typically cerebellum and vermis are affected equally, our case, like patient 7 of Moog [4] and patients 10, 11 and 12 of Burglen [8] had a subtly affected vermis. CASK gene duplication, found in our case, has been reported only in a small number of reported MICPCH syndrome cases [3],[4] and therefore, seems to be a less common mutational mechanism to produce MICHPCH syndrome. Despite extensive investigations in our case, the diagnosis remained elusive until a-CGH analysis uncovered micro-duplication, which was earlier reported in two cases by Hayashi [7] and in patients 21 and 25 reported by Moog. [4] Based on the preceding discussion, it can be suggested that in girls presenting with severe neurodevelopmental delay and microcephaly, if neuroimaging depicts ponto-cerebellar hypoplasia with normal CC, a-CGH analysis may be carried out to ascertain genetic diagnosis. [2],[3],[4],[5],[8]
| Conclusion | | |
MICPCH syndrome, mostly seen in girls, is caused by de novo loss-of-function CASK gene mutations in X-chromosome in the Xp11.4 region, producing null alleles. Our case exemplifies the typical clinico-radiologic phenotype linked to CASK gene mutations. Clinicians assessing girls presenting with severe developmental delay and microcephaly can use the radiological features of ponto-cerebellar hypoplasia with normal corpus callosum and low cerebrum/CC ratio as a guide to look for CASK gene mutations by using a-CGH analysis.
| References | | |
| 1. | Shinawi M, Cheung SW. The array CGH and its clinical applications. Drug Discov Today 2008;13:760-70. 
|
| 2. | Najm J, Horn D, Wimplinger I, Golden JA, Chizhikov VV, Sudi J, et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat Genet 2008;40:1065-7.
|
| 3. | Hayashi S, Mizuno S, Migita O, Okuyama T, Makita Y, Hata A, et al. The CASK gene harbored in a deletion detected by array-CGH as a potential candidate for a gene causative of X-linked dominant mental retardation. Am J Med Genet A 2008;146A: 2145-51.
|
| 4. | Moog U, Kutsche K, Kortüm F, Chilian B, Bierhals T, Apeshiotis N, et al. Phenotypic spectrum associated with CASK loss-of-function mutations. J Med Genet 2011;48:741-51.
|
| 5. | Takanashi J, Arai H, Nabatame S, Hirai S, Hayashi S, Inazawa J, et al. Neuroradiologic features of CASK mutations. AJNR Am J Neuroradiol 2010;31:1619-22.
|
| 6. | Froyen G, Van Esch H, Bauters M, Hollanders K, Frints SG, Vermeesch JR, et al. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: Important role for increased gene dosage of XLMR genes. Hum Mutat 2007;28:1034-42.
|
| 7. | Hayashi S, Okamoto N, Chinen Y, Takanashi J, Makita Y, Hata A, et al. Novel intragenic duplications and mutations of CASK in patients with mental retardation and microcephaly with pontine and cerebellar hypoplasia (MICPCH). Hum Genet 2012;131:99-110.
|
| 8. | Burglen L, Chantot-Bastaraud S, Garel C, Milh M, Touraine R, Zanni G, et al. Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: Confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet J Rare Dis 2012;7:18.
|
| 9. | Zheng CY, Seabold GK, Horak M, Petralia RS. MAGUKs, synaptic development, and synaptic plasticity. Neuroscientist 2011;17:493-512.
|
| 10. | Hsueh YP. The role of the MAGUK protein CASK in neural development and synaptic function. Curr Med Chem 2006;13:1915-27.
|
| 11. | Hsueh YP. Calcium/calmodulin-dependent serine protein kinase and mental retardation. Ann Neurol 2009;66:438-43.
|
[Figure 1]
|









